Keywords
MGISEQ-2000, DNBSEQ-G400, NGS, Paired-end sequencing, fastq merging
This article is included in the Cell & Molecular Biology gateway.
MGISEQ-2000, DNBSEQ-G400, NGS, Paired-end sequencing, fastq merging
No specific changes to the uploaded data, or affiliation or names. Some explanations are added in the conclusion section accordingly to the reviewer's comment. We pointed out that the proposed script provides merging of reads, but does not guarantee the good quality of the received data. In this case, the researcher must decide whether to restore the data or not, depending on the emergency situation, and be sure to check the data quality for in-lab reference samples.
See the authors' detailed response to the review by Sergey Knyazev
At the end of 2017, Chinese company MGI Tech presented the MGISEQ-2000 sequencing platform1, promoting it as a device for large and medium scale genome sequencing. MGISEQ is specific in harnessing cPAS sequencing technology and using nanoballs (DNB) generated from circular molecules of DNA library by rolling circle replication2. MGISEQ is compatible with a wide range of reagents for sequencing in SE50, SE100, SE400, PE100, PE150, and PE200 modes. MGISEQ-2000 provides the quality of sequencing comparable with that of the Illumina platform3–6.
The first MGISEQ-2000 sequencer in Russia was installed in our lab (Center for Precision Genome Editing and Genetic Technologies for Biomedicine, Pirogov Medical University) in February 2019, and we run it once a week in the paired-end 150 mode (PE150). According to our experience, one PE150 run usually takes 68 hours if one flow cell is used at a time. During one of these runs, about 23.00 on Saturday, there was a failure of the Moscow power grid leading to a 50-minute blackout of a whole district including Pirogov Medical University. UPS battery storage was sufficient only for 20 extra minutes, then the sequencer turned off until the power was restored. Therefore, the device with loaded reagents remained in sleep mode for 35 hours until Monday. Before the instrument was switched off, it had performed 138 full cycles of forward read sequencing (run 27). The specific feature of the MGISEQ-2000 sequencing program is that it reads a barcode at the end of a run after it completes sequencing of forward and reverse reads. So, to a first approximation, the data obtained could not be demultiplexed as information on the barcodes was absent.
According to the MGI Tech7 recommendations, after consulting with the MGI Tech service engineers, we were advised to dispose of the current tank with reagents as well as the flow cell and run the samples using new reagents. In the first place, it is linked to the high sensitivity of the MDA reagent to storage at +4°C as it loses its activity very quickly. We decided to continue the run using the reagents that had been loaded for the weekend and try to restore the data. Finally, we managed to rescue the data using the software from ZebraCall8 and our own script on C++, which is reported here https://github.com/genomecenter/runcer-necromancer.
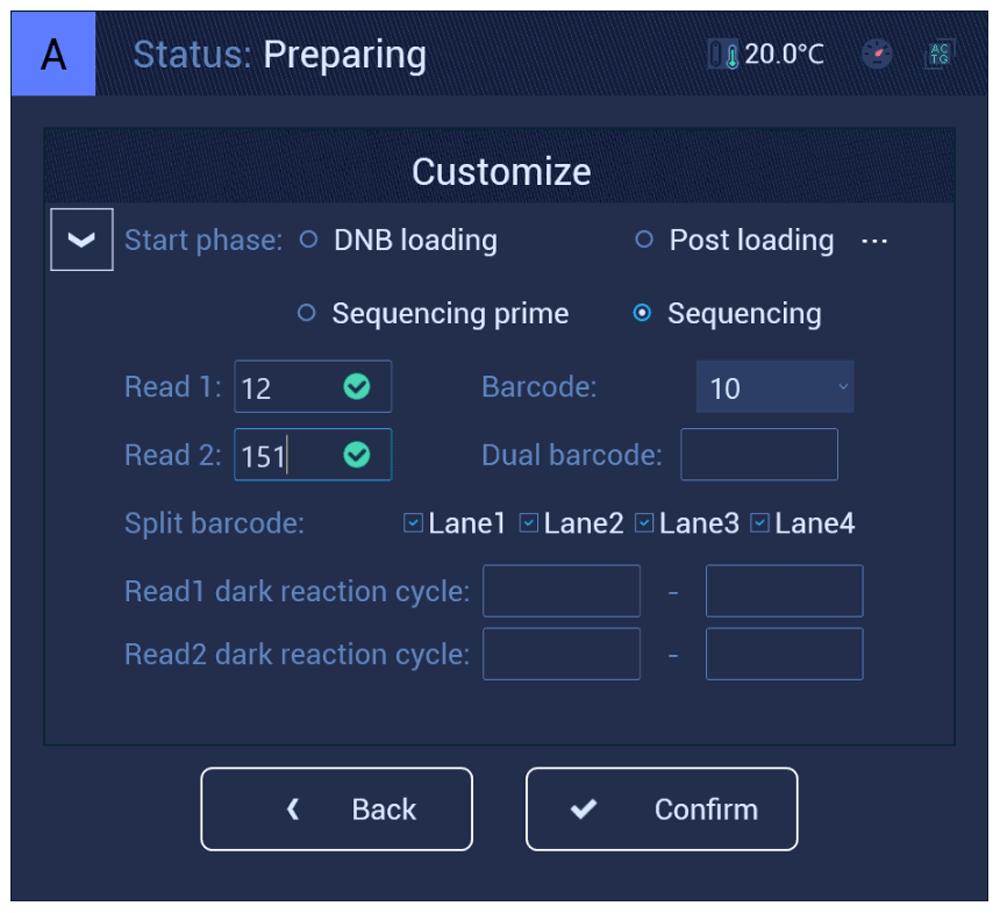
We prepared 3 pools of circularized libraries following the standard MGI Tech protocol9. Then we synthesized DNB, loaded a flow cell using the MGIDL-200H manual loader, prepared a sequencing cartridge from the MGISEQ-2000RS High-throughput Sequencing Set with User manual version: A2, and started sequencing on A-side in PE150 mode. Run 27 was aborted at the 139th cycle of the read-1 sequencing phase. After 35 h, we restarted the run (run 27_2) using the same sequencing cartridge and flow cell in a custom mode with the following parameters: read 1 for 12 cycles, read 2 for 151 cycles, Start phase: Sequencing (Figure 1). For summary reports generated by MGISEQ-2000 for lane 1 of runs 27 and 27_2, see Extended data: File S1, S2.
The generation of .fastq files containing forward reads for the interrupted run was performed using ZebraCall v28 framework (C\:ZebraCallV2\client.exe – the pathway to software on MGISEQ-2000), which transforms intermediate .cal files into fastq format and demultiplexes them using barcodes.
The appropriate work of ZebraCall requires a .txt file with barcode sequences used for demultiplexing. We created an empty file 'empty_barcode.txt' so that the last 10 nucleotides from 13 nucleotides that were read earlier would not be recognized as barcodes by ZebraCall.
We used the following command (we provide an example for lane 1):
client.exe D:\Result\workspace\run_name\L01 139 6 72 -B C:\ZebraCallV2\empty_barcode.txt -N run_name -U 1 -FIt contains the options:
the access to the folder with .cal files
run_name — the name of a run
139 – the number of completed sequencing cycles
6 72 – the number of fields of view counted horizontally and vertically for a corresponding lane
-B – a path to the file with barcodes
-U – the number of a lane
-F – fastq generation without generation of flow cell images
As a result, for each lane, we generated files 'run_name_L0N_read.fq.gz' where N is a lane number. Such file contained a read name and a sequence of 138 nucleotides long.
MGISEQ-2000 employs a patterned flow cell, so each DNB in a cell has unique coordinates at X and Y axes which do not depend on flow cell localization in a device and are not changed if the flow cell is displaced. When the power of the sequencer was off, the vacuum pump was switched off as well. The coordinates of each read were saved in a header of a .fastq file (Figure 2). This allowed us to integrate the data on forward reads obtained before and after the instrument was off.
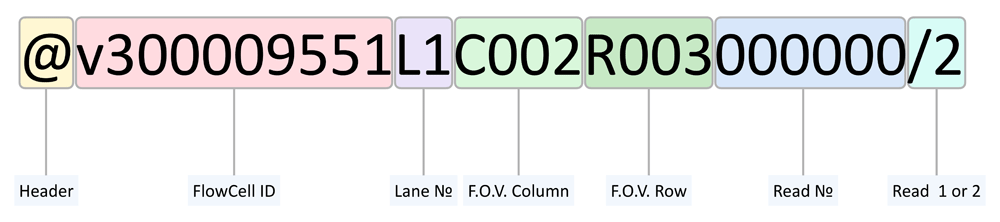
As a read number being used for forward and reverse reading is unique, we managed to combine the 138-nucleotide sequences obtained during the first run with the nucleotide sequences obtained during the second run based on the information on F.O.V Column, F.O.V Row, and read numbers. To achieve this, we created a C++ script, which can be accessed at GitHub https://github.com/genomecenter/runcer-necromancer. The instruction for script running can be found below and in the file README.md in the repository.
The script (http://doi.org/10.5281/zenodo.431680210) is executable on Linux (was tested on Ubuntu 20.04) with GCC compiler with C++17 support and zlib (apt-get install zlib1g-dev). First step is a building: you need to run build.sh script inside the root folder. SaveReads program recovers sample files by placing fixed files into the fixed directory inside current directory. It is important to check that there are no identical filenames between samples files. SaveReads accepts N+1 argument, where first argument is _undecoded.fq.gz (pool of non-demultiplexed reads) file from interrupted run, and next N arguments are standard samples files. Script SaveReads.py simplifies call to SaveReads. This file accepts pool of non-demultiplexed reads as its single argument. All files with _1.fq.gz ending from current folder will be taken as samples files.
The most important parameter for sequencing quality is the ratio of the data with the quality level of no less than Q30. The Q30 value and other quality metrics did not decrease dramatically in spite of a 35-hour stand by (Figure 3, Table 1).
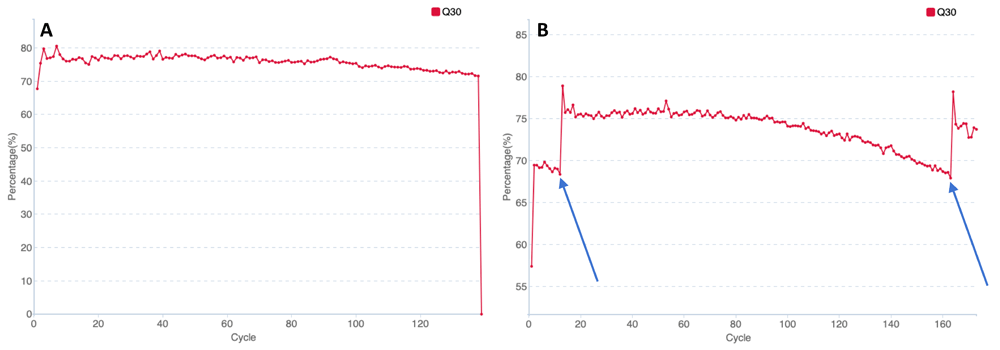
Q30 histograms for runs 27 (А) and 27_2 (B). The X-axis represents the number of sequencing cycles, the Y-axis represents the ratio of the data with the quality no less than Q30 (%). Blue arrows in histogram B indicate the cycles which reverse reading and barcode reading started.
| Run | 27 | 27_2 | Drop |
|---|---|---|---|
| Loaded DNBs (from the first base report) | 1169312 | 1131610 | 3% |
| Chip productivity, % | 76.82 | 68.18 | 11% |
| Q30, % | 90 | 90.96 | 0% |
| Total reads per lane, M | 467.3 | 397.67 | 15% |
This implies that storing a loaded cartridge for 35 h leads to its decline, however, it can be still used for sequencing.
To check if merging reads from different runs was correct, we compared 7 samples of whole-exome sequencing from runs 27 and 27_2 with the data from the same samples obtained in run 22. We used the distribution of the size of an insert between left and right reads and the ratio of reads having the insert size exceeding 1000 nucleotides as control metrics. If read merging had been performed with errors, the portion of the reads mapped to various genome regions would have significantly increased. See Figure 4 for the distribution of an insert size for sample LWX777 from the control group.
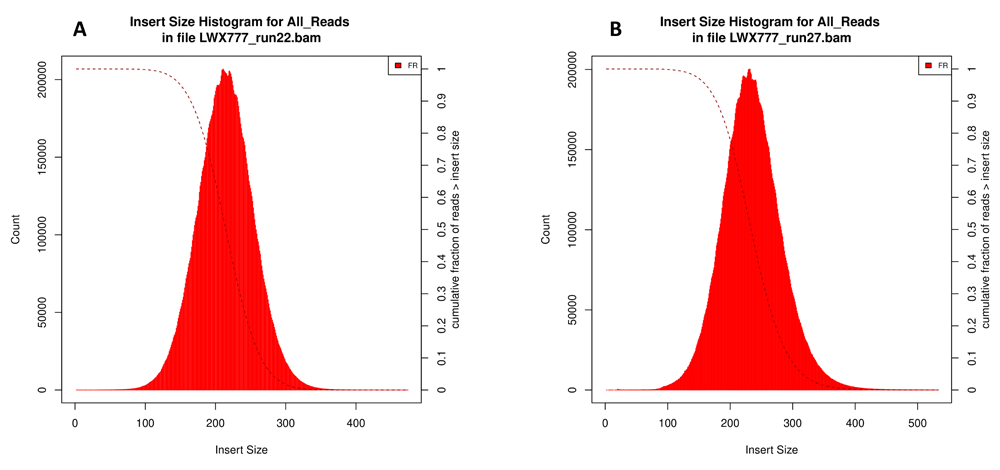
The distribution of inserts for sample LWX777 from runs 22 (A) and 27+27_2 (B). The X-axis represents the number of reads, the Y-axis represents an insert size. The diagrams were obtained using Picard CollectIsertSizeMetrics v2.22.4.
The ratio of reads from sample LWX777 with the insert size exceeding 1000 nucleotides was 0.003% in case of the data combined from the different runs, while it was 0.005% in case of the previous sequencing without read integration. The obtained data imply that read merging was correct.
It is possible to use a sequence cartridge after 35-hour storage at +4°C, although the quality of the obtained data is reduced.
We would like to point out that the method we propose can be applied in exceptional cases at your own risk and always followed by a quality check of the obtained data. As the procedure described above is not a complete experiment with replication and control samples, we cannot guarantee the quality of the data after merging reads of sequencing run under different problematic conditions. Therefore, we recommend that researchers in such situations adequately assess the conditions: how long a sequencing run was interrupted (hours or days), how the temperature and humidity in the laboratory room and inside the device changed, at which stage of sequencing (reading forward or reverse read, MDA reaction, reading a barcode) the power was turned off, etc. It is also desirable to use an in-lab reference sample in each run of the instrument to assess data quality and batch-effect. The described aborted run had a DNA library sample that we had previously sequenced under normal conditions, so we were able to assess the quality of the data obtained after merging reads in terms of GC-content, Ti/Tv and het/hom ratio, coverage statistics, variant calling.
Merging sequencing data can be successfully performed if the information about the localisation in flow cells is saved in a read header. The researcher must then compare the data quality on their own reference samples to decide whether to use the data from the aborted run.
Raw data for the sample LWX777 from runs 22 and 27_1+27_2 available at Sequence Read Archive (SRA), BioProject ID PRJNA683755: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA683755/
LWX777_run27_2_united (SRA: SRS7871577) is an example of reconstructed fastq file from the 2 parts of interrupted run 27. First 139 nucleotides were received form run_name_L04_read.fq.gz fastq file with non-demultiplexed left reads for lane 4. LWX777_run22 (SRA: SRS7871575) is fastq files from previous run 22.
Zenodo: genomecenter/runcer-necromancer: Runcer Necromancer updated release (December 2020), http://doi.org/10.5281/zenodo.434035010.
This project contains the following extended data:
File S1. Summary report for run 27 lane 1 from MGISEQ-2000
File S2. Summary report for run 27_2 lane 1 from MGISEQ-2000
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
Script available from: https://github.com/genomecenter/runcer-necromancer
Archived script as at time of publication: http://doi.org/10.5281/zenodo.434035010.
License: MIT
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)