Keywords
Miscanthus, biofuel, C4, assembly, annotation
This article is included in the Plant Science gateway.
This article is included in the Genomics and Genetics gateway.
Miscanthus, biofuel, C4, assembly, annotation
Miscanthus is a genus of C4 perennial rhizomatous grasses native to East Asia and Oceania, and naturally adapted to a wide range of climate zones and land types. Miscanthus sacchariflorus is among the most widely distributed species within the genus. It originated in the Yellow Sea region of China and can be predominantly found in cool latitudes of East Asia with varying ploidy1. M. sacchariflorus occurs in both diploid (2n = 38) and tetraploid (2n = 76) forms, where tetraploid M. sacchariflorus genotypes originated by autopolyploidy2. M. sacchariflorus probably has the greatest winter hardiness among all the Saccharinae3.
Natural interspecific Miscanthus hybrids are commonly observed, even between individuals of different ploidy. For example, introgression of M. sacchariflorus is often found among cultivated European M. sinensis ecotypes1,4. Furthermore, M. x giganteus, a sterile triploid hybrid resulting from the hybridization between M. sinensis and M. sacchariflorus, is the predominant commercially grown species owing to its high biomass productivity and low chemical input requirements. The common occurrence of hybridization events and variable ploidy are challenging to the improvement of these bioenergy grasses and increase the need for genomic resources from different Miscanthus species. A chromosomal-scale reference genome using a doubled-haploid M. sinensis line was recently published4.
We assembled, annotated and validated a draft genome from the diploid M. sacchariflorus cv. “Robustus 297” genotype, as well as generating rhizome, stem and leaf RNA-Seq data from the same genotype. This dataset was previously used to verify that both M. sinensis and M. sacchariflorus share the same A/B ancestral tetraploidy4. Here, we present the first draft genome of M. sacchariflorus, the second Miscanthus genome available after M. sinensis4.
DNA was extracted from leaves from the diploid M. sacchariflorus cv. “Robustus 297” genotype (Biosample SAMN08580354) using the Qiagen DNeasy kit. RNA was also extracted from leaf, stem and root tissues from the same plant. All samples were taken from a plant grown from seed in trays in a glasshouse in 2009. This genotype is established and used in breeding at IBERS (Wales, UK). The RNA-seq libraries were deposited as part of previous work in the BioProject PRJNA639832.
We obtained ~5.86e9 pairs of 100 bp paired-end reads from an Illumina paired-end library with a 560 bp insert-size that was sequenced on Illumina HiSeq 2500 machines in rapid run mode by the Earlham Institute. This represents approximately 50X coverage of the heterozygous content and 100X coverage of the homozygous content of the genome. Read quality was assessed, and contaminants and adaptors removed using Kontaminant5. These paired-end short-reads were assembled into 17M contigs with a total length of 3.27 Gb using ABySS6 version 1.5.1, with default options and a kmer size of 71.
We obtained ~141.1e6 pairs of reads from a Nextera 150 bp mate-pair library with approximately 7 Kb insert-size, which was used for scaffolding the previous contigs together with the paired-end reads, using SSPACE7 without “extension” step. Nextera mate-pair reads were required to include a fragment of the adaptor to be used in the scaffolding step5, and we filtered out sequences shorter than 500 bp. We obtained 589K scaffolds, a total length of 2.54 Gb with an N50 of 10.2 Kb. This whole-genome assembly was denominated “Msac_v2” and is deposited at NCBI in BioProject PRJNA679435.
Our gene structure annotation pipeline8 used five sources of evidence that were provided to AUGUSTUS9 (version 2.7) for gene annotation: (1) Repetitive and low complexity regions of the scaffolds identified using RepeatMasker10 (version open-4.0.5) based on homology with the RepBase11 public database (Release 20140131) and a new database of repeat elements identified in the assembly with RepeatModeler12. The repeats annotation was deposited in Zenodo (See data availability); (2) exon-intron junctions identified by Tophat13 (version 2.1.0); (3) de novo and genome-guided ab initio transcripts assembled with Trinity14 (version 2.6.5 )and Cufflinks15 (version 2.2.1) from RNA-Seq reads obtained from several tissues from the same genotype; (4) ab initio gene models predicted by SNAP16 (version 29-11-2013) and GeneID17 (version 1.4.4); and (5) homology-based alignments of transcripts and proteins from Miscanthus sinensis and maize using Exonerate18 with a minimal identity of 0.7 and coverage of 0.7. Finally, AUGUSTUS9 was run with the options “genemodel=complete” and “alternatives-from-evidence=true” to ensure that the predicted genes were compatible with all the previous provided evidence.
For the functional annotation of these predicted genes, translated gene sequences were compared with the NCBI non-redundant (nr 20170116) proteins and EBI’s InterPro (version 5.22.61) databases, and the results were imported into Blast2GO19 to annotate the GO and GO slim terms, enzymatic protein codes and KEGG pathways. A similar GO annotation from translated gene sequences can be done with eggNOG-mapper20. These functional descriptors were deposited in Zenodo (See Underlying data).
To improve the genome contiguity, we anchored our M. sacchariflorus scaffolds to the Miscanthus sinensis genome4. However, no nucleotide content from M. sinensis was incorporated in the M. sacchariflorus assemblies.
Firstly, scaffolds longer than 2 kbps from the whole genome assembly “Msac_v2” were scaffolded again using SSPACE7 and the M. sinensis mate-pairs reads, the gaps between scaffolds were filled in with Ns. This new whole-genome assembly was denominated “Msac_v3”, and was deposited at NCBI in Bioproject PRJNA435476, under the GenBank accession GCA_002993905. It contains 137,916 scaffolds for a total of 2.074 Gb with an N50 of 25.6 Kbps. The gene annotation was projected to the “Msac_v3” assembly using PASA21 (version 2.0.1): genes were aligned to the new assembly using GMAP, requiring a minimum identity of 0.85 and coverage of 0.55, and later validated using the default parameters in PASA.
Finally, we obtained the chromosomal position in the M. sinensis chromosomes of the scaffolds from the “Msac_v3” assembly. Using Satsuma222 (version untagged-330e3341a1151a978b37), we identified every perfect-identify match between both assemblies (3,635,504 matches in total). The coordinates of these matches in BED 8 format were used as input to the “OrderOrientBySynteny” script from Satsuma2, which identifies the best chromosomal position for each scaffold. These position coordinates are available as an AGP file as part of GCA_002993905, which anchors our final whole-genome assembly to 19 chromosomes (accessions CM00959 to CM009609 in NCBI).
RNA-seq cleaned reads from each tissue were independently aligned to both assembly versions using STAR23 (version 2.6.0c). BUSCO24 (version 4.1.4) was used to assess completeness with the single-copy orthologs database for green plants (Viridiplantae, version 2020-09-10). Orthologs were identified using Orthofinder225 (version 2.3.12) with default parameters and the option “-msa”, which directly provided comprehensive statistics comparing the provided proteomes. All the proteomes from the other species used (Table 1) were downloaded from Phytozome (v7.1 DOE-JGI). Genomes were aligned using Minimap226 (version 2.17) with the “asm10” parameter for related genomes, secondary alignments (tp:A:S) filtered out, and results visualised using dotPlotly27 (Github version, latest updated on 4 May 2018).
| Msac_v2 (unanchored) | Msac_v3 (anchored by M. sinensis) | Reference: M. sinensis4. | |
|---|---|---|---|
| NCBI bioproject | PRJNA679435 | PRJNA435476 (GCA_002993905) | v.7.1 from Phytozome |
| Length | 2.539 Gb | 2.074 Gb | 1.68 Gbps |
| Scaffolds | 588,758 scaffolds | 137,931 scaffolds* | 19 Chrs and 14,414 scaffolds |
| N20 | 25.39 Kbps | 62.61 Kbps | 146.1 Mbps |
| N50 | 10.25 Kbps | 25.63 Kbps | 88.51 Mbps |
| N80 | 2.79 Kbps | 9.42 Kbps | 75.06 Mbps |
| Max | 378.48 Kbps | 458.83 Kbps | 160.9 Mbps |
| ANNOTATION | Msac_v2 | Msac_v3 | M. sinensis |
| Gene models | 81,431 | 68,578 | 67,967 |
| Proteins | 86,767 | 68,578** | 67,789 |
| BUSCO | Msac_v2 | Msac_v3 | M. sinensis |
| Complete | 55.5% (48% in single copy) | 59.8% (50.4% in single copy) | 97.6% (36.2% in single copy) |
| Fragmented | 32.2% | 26.4% | 1.6% |
| Missing | 12.3% | 13.6% | 0.8% |
| RNA MAPPING | Msac_v2 | Msac_v3 | M. sinensis |
| Reads mapping in the genome once (root, stem and leaf) | 76.2% 76.4% 78.8% | 75% 76.7% 78.1% | 78.8%*** 83.5% 82.5% |
| Reads mapping in the genome multiple times (root, stem and leaf) | 22.5% 23% 20.7% | 19.5% 18.8% 17.3% | 19.7%*** 15.5% 16.6% |
We produced two whole-genome assemblies for M. sacchariflorus that we named “Msac_v2” and “Msac_v3”, with total lengths of 2.54 Gbps and 2.074 Gbps, respectively (Table 1). The difference in size is mainly a result of filtering 402 Mb from sequences under 2 kb in the latter before anchoring to the M. sinensis genome. Our “Msac_v2” assembly covered ~59 % of M. sacchariflorus genome size, which is estimated to be 4.3 Gb28. Approximately 40% of the assembly was composed by transposable elements (987.3 Mb; Table 2), including 491 Mb (19.4%) and 154 Mb (6.1%) by copies of the Gypsy and Copia LTRs, respectively; and 180 Mb (7.1%) by several class 2 DNA transposons (MULE, CMC, Harbinger, etc.)
We identified 219,394 primary alignments longer than 2 kb between the unanchored M. sacchariflorus (“Msac_v2”) and M. sinensis. The resulting dotplot (Figure 1) shows the conserved synteny between both species, which diverged 1.6 Mya4. Figure 1 also shows the highly conserved synteny between the pairs of homoeologous chromosomes (e.g. green boxes in chromosomes one and two), and the fusion in chromosome 7 of the chromosome homeolog to chromosome 13; which was also reported in M. sinensis4. There are several large inversions between chromosomes 9 and 10, and 3 and 4 (cyan boxes in Figure 1). Our assembly of a heterozygous genotype resulted in multiple heterotigs (heterozygous contigs) containing the alternative or secondary haplotypes (e.g. pink boxes in Figure 1).
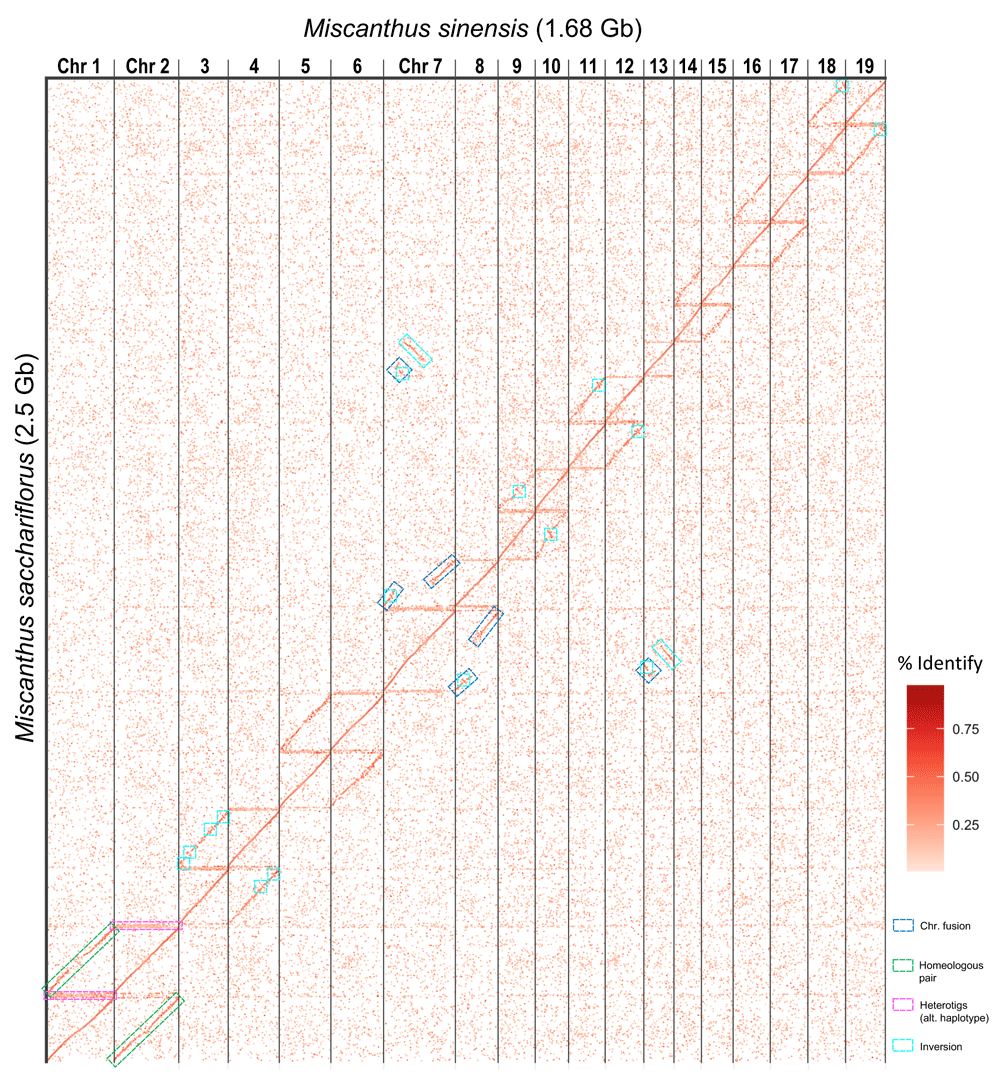
The plot shows the primary alignments longer than 2 kbps between both species. The M. sacchariflorus scaffolds (Y-axis) have been sorted by their coordinates in M. sinensis chromosomes (X-axis). Large homoeologous blocks and chromosomal rearrangements are highlighted in boxes.
The utility of our assemblies for genomic studies is evidenced by the proportion of RNA-seq from three different tissues from the same M. sacchariflorus genotype that aligned to the assemblies. On average 99% and 95% of the RNA-seq reads aligned in “Msac_v2” and “Msac_v3”, respectively (Table 1).
We estimated that we assembled more than 85% of the M. sacchariflorus genes. Furthermore, our assemblies include several alleles of genes in the heterozygous regions of the genome, while the M. sinensis reference was generated from a double-haplotyped genotype. The estimation of the proportion of assembled genes (~85%) was supported by (1) the results from BUSCO, which reported 86.4–87.7% of presented core genes, of which ~2/3rds were complete (Table 1); and (2) the difference in the number of proteins from related species for which we can identify an ortholog in M. sacchariflorus compared to M. sinensis, as control, using Orthofinder2 (Table 3).
Based on the results from Orthofinder2 (Table 3), we found orthologs in M. sacchariflorus for 64.5% of the M.sinensis annotated proteins, so we estimated ~1/3rd of the Miscanthus proteins to be specific to each species. On the other hand, we estimated that ~3,000 genes may be missing in the “Msac_v2” annotation based on the number of Sorghum bicolor proteins with orthologues in M. sinensis but absent in M. sacchariflorus. Better estimations were obtained with the other four species, where the genes absent in Msac_v2 compared with M. sinensis were estimated to be 254, 579 and 1627 (Table 3). Additionally, ~6,000 genes could be missed in “Msac_v3” compared to “Msac_v2” based on the difference in the number of M. sinensis orthologues in each assembly (Table 3). This is likely from genes in the sequences shorter than 2 Kbps (totalling 402 Mbps) that were filtered out before anchoring. There was a large difference in the proportion of “fragmented” BUSCO genes found in the M. sacchariflorus (32.2%) and M. sinensis (1.6%) assemblies (Table 1). To assess if that difference had an effect on the quality of the annotation, we compared the number of proteins from M. sacchariflorus and M. sinensis for which we can identify an ortholog in another species (Table 3); we found the difference between both Miscanthus species ranged between 6,571 proteins when compared to sorghum (43,475 to 37,219; Table 2) to only 121 when compared to maize (39,986 to 38,478, Table 3).
In conclusion, our M. sacchariflorus genome can served as the basis for functional genetic analyses on Miscanthus, one of the main biofuel grass crops used in temperate latitudes. However, there are opportunities to improve it using new approaches, such as long-reads.
NCBI BioProject: Miscanthus sacchariflorus cultivar:Robustus 297. Accession number PRJNA435476; https://identifiers.org/bioproject:PRJNA435476.
This BioProject contains the raw paired-end and mate-pair reads.
NCBI BioProject: RNA-seq Miscanthus hybrids with contrasting phenotypes. Accession number PRJNA639832; https://identifiers.org/bioproject:PRJNA639832.
This BioProject contains RNA-seq reads, deposited as part of a previous project29.
NCBI BioProject: Miscanthus sacchariflorus cultivar:Robustus 297. Accession number PRJNA679435; https://identifiers.org/bioproject:PRJNA679435.
This Bioproject contains the unanchored “Msac_v2” assemblies and gene annotations under accession JADQCR000000000.
The anchored “Msac_v3” assemblies and gene annotations are deposited under accession accession GCA_002993905 under Bioproject PRJNA435476.
The chromosomal positions in the M. sinensis chromosomes of the scaffolds from the “Msac_v3” assembly are available in an AGP file as part of GCA_002993905, which places the scaffolds in 19 chromosomes (accessions CM009591 to CM009609 in NCBI).
Zenodo: Supplementary dataset to "Draft genome assembly of the biofuel grass crop Miscanthus sacchariflorus". http://doi.org/10.5281/zenodo.4270235.
This project contains the assemblies in FASTA format, gene annotations in GFF3 format, functional annotations in tabulated text format, and AGP file with anchoring information.
Data deposited with Zenodo are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)