Keywords
software comparison, bioinformatics tool, lysogen genome, temperate phage, prokaryotic virus
This article is included in the Bioinformatics gateway.
software comparison, bioinformatics tool, lysogen genome, temperate phage, prokaryotic virus
In this version we address the comments of the reviewers. The comparison now includes two additional programs (ProphET and Seeker), and an additional 29 manually-curated genomes to the gold-standard library; we have updated the results accordingly. We compare f1 scores of the different genera that are present in the gold-standard library to assess how the performance of the programs may be influenced by taxonomy. The underlying data now includes our guidelines for manually curating genomes with prophage annotations.
See the authors' detailed response to the review by Karthik Anantharaman and Kristopher Kieft
See the authors' detailed response to the review by Franklin Nobrega
Bacteriophages (phages), viruses that infect bacteria, can be either temperate or virulent. Temperate phages may integrate into their bacterial host genome. Such integrated phage genomes are referred to as prophages and may constitute as much as 20 percent of bacterial DNA (Casjens, 2003). They replicate as part of the host genomes until external conditions trigger a transition into the virulent lytic cycle, resulting in replication and packaging of phages and typically the death of the host bacteria. Prophages generally contain a set of core genes with a conserved gene order that facilitate integration into the host genome, assembly of phage structural components, replication, and lysis of the host cell (Kang et al., 2017; Canchaya et al., 2003). As well as these core genes, phages can contain an array of accessory metabolic genes that can effect significant phenotypic changes in the host bacteria (Breitbart, 2012). For instance, many prophages encode virulence factors such as toxins, or fitness factors such as nutrient uptake systems (Brüssow et al., 2004). Lastly, many prophages encode a variety of super-infection exclusion mechanisms to prevent concurrent phage infections, including restriction/modification systems, toxin/antitoxin genes, repressors, etc. reviewed in Abedon (2015, 2019). The function of most prophage accessory genes remains unknown.
Core (pro) phage genes have long been used for identifying prophage regions. However, there are other unique characteristics that can distinguish prophages from their host genomes: bacterial genomes have a GC skew that correlates with the direction of replication, and the insertion of prophages will generally disrupt this GC bias (Grigoriev, 1998). Transcript direction (Campbell, 2002) and length of prophage proteins have also proven to be useful metrics in predicting prophages (Akhter et al., 2012; Song et al., 2019), where phage genes are generally smaller and are oriented in the same direction (Dutilh et al., 2014). Likewise, gene density tends to be higher in phage genomes and intergenic space shorter (Amgarten et al., 2018; McNair et al., 2019).
Over the last two decades many prophage prediction tools have been developed, and they fall into two broad classes: (1) web-based tools where users upload a bacterial genome and retrieve prophage annotations including PHASTER (Arndt et al., 2016), Prophage Hunter (Song et al., 2019), Prophinder (Lima-Mendez et al., 2008), PhageWeb (Sousa et al., 2018), and RAST (Aziz et al., 2008); and (2) command-line tools where users download a program and database to run the predictions locally (although some of these also provide a web interface for remote execution). In this work we focus on this latter set of tools (Table 1) because web-based tools typically do not handle the large numbers of simultaneous requests required to run comparisons across many genomes.
| Tool (year) | Version | Package manager | Dependencies | Database size | Approach | Citation |
|---|---|---|---|---|---|---|
| Phage Finder (2006) | 2.1 | Aragorn, BLAST-legacy, HMMer, Infernal, MUMmer, tRNAscan-SE | 93 MB | Legacy-BLAST, HMMs | (Fouts, 2006) | |
| PhiSpy (2012) | 4.2.6 | conda, pip | Python3, BioPython, NumPy, SciPy | 47 MB required, 733 MB optional (pVOGs) | Gene and nucleotide metrics, AT/CG skew, kmer comparison, machine learning, HMMs, annotation keywords | (Akhter et al., 2012) |
| VirSorter (2015) | 1.0.6 | conda | MCL, Muscle, BLAST+, BioPerl, HMMer, Diamond, Metagene_annotator | 13 GB | Alignments, HMMs | (Roux et al., 2015) |
| ProphET (2019) | 0.5.1 | BLAST-legacy, EMBOSS, BedTools, Perl, BioPerl | 41 Mb | Legacy-BLAST searches | (Reis-Cunha et al., 2019) | |
| Phigaro (2020) | 2.3.0 | conda, pip | Python3, Beautifulsoup4, BioPython, bs4, HMMer, lxml, NumPy, Pandas, Plotly, Prodigal, PyYAML, shsix | 1.6 GB | HMMs | (Starikova et al., 2020) |
| DBSCAN-SWA (2020) | 2e61b95 | Numpy, BioPython, Scikit-learn, Prokka | 2.2 GB | Gene metrics, alignments | (Gan et al., 2020) | |
| VIBRANT (2020) | 1.2.1 | conda | Python3, Prodigal, HMMer, BioPython, Pandas, Matplotlib, Seaborn, Numpy, Scikit-learn, Pickle | 11 GB | HMMs (KEGG, Pfam, VOG), machine learning | (Kieft et al., 2020) |
| Seeker (2020) | 1.0.3 | pip | Python3, TensorFlow | 64 kb | Machine learning (LSTM) | (Auslander et al., 2020) |
| PhageBoost (2021) | 0.1.7 | pip | Python3 | 13 MB | Gene and nucleotide metrics, machine learning | (Sirén et al., 2021) |
| VirSorter2 (2021) | 2.2.1 | conda | Python3, Snakemake, Scikit-learn, imbalanced-learn, Pandas, Seaborn, HMMer, Prodigal, screed | 12 GB | Alignments, HMMs | (Guo et al., 2021) |
Despite the abundance of prophage prediction algorithms, there has never been either a set of reference genomes against which all tools can be compared, nor a unified framework for comparing those tools to identify their relative strengths and weaknesses or to identify opportunities for improvement. We generated a set of manually annotated bacterial genomes released under the FAIR principles (Findable, Accessible, Interoperable, and Reusable), and developed an openly available and accessible framework to compare prophage prediction tools.
To assess the accuracy of the different prophage prediction tools, a set of 57 gold-standard publicly available bacterial genomes with manually curated prophage annotations was generated. We combined this with the 21 manually annotated genomes described in Casjens (2003) that were not already included for a total of 78 genomes for evaluating the bioinformatics tools. The genomes and prophage annotations currently included are available in Table S1. The genomes are in GenBank format and file conversion scripts are included in the framework to convert those files to formats used by the different software. The tools that are currently included in the framework are outlined in Table 1. Snakemake (Köster & Rahmann, 2012) pipelines utilising conda (Anaconda Software Distribution. Conda. v4.10.1, April 2021 (Conda, RRID:SCR_018317)) package manager environments were created for each tool to handle the installation of the tool and its dependencies, running of the analyses, output file conversion to a standardized format, and benchmarking of the run stage. Where possible, gene annotations from the GenBank files were used in the analysis to promote consistency between comparisons. DBSCAN-SWA was not able to consistently finish when using GenBank files as input, and instead the genome files in fasta format were used. Another pipeline was created to pool the results from each tool and some comparisons are illustrated in the included Jupyter notebook. Testing and development of the pipelines were conducted on Flinders University’s DeepThought HPC infrastructure. The final benchmarking analysis was performed on a stand-alone node consisting of dual Intel® Xeon® Gold 6242R processors (40 cores, 80 threads), 768 GB of RAM, and 58 TB of disk space. Each tool was executed on all genomes in parallel (one thread per job), with no other jobs running. The only exception to this was Seeker which was run one at a time on a single core due to high memory requirements (see below).
The runtime and CPU time in seconds, peak memory usage and file write operations were captured by Snakemake (Snakemake, RRID:SCR_003475) for the steps running the prophage tools only (not for any file conversion steps before or after running each tool). The predictions were compared to the gold standard prophage annotations and the number of true positive (TP), true negative (TN), false positive (FP) and false negative (FN) gene labels were used to calculate the performance metrics. Each application marks prophages slightly differently, and therefore we used the designation of coding sequence (CDS) features as phage or not to assess prophage predictions.
We developed the framework to simplify the addition of new genomes to the benchmarks. Each genome is provided in the standard GenBank format, and the prophages are marked by the inclusion of a non-standard flag for each genomic feature that indicates that it is part of a prophage. We use the qualifier/is_phage=“1” to indicate prophage regions. Our guidelines for manually annotating prophages in bacterial genomes are available in the GitHub repository at github.com/linsalrob/ProphagePredictionComparisons/blob/master/Supplementary/prophageAnnotation.md.
We compared the availability, installation, and results from ten different prophage prediction algorithms (Table 1). LysoPhD (Niu et al., 2019) could not be successfully installed and was not included in the current framework (see below). The remaining ten—PhiSpy (Akhter et al., 2012), Phage Finder (Fouts, 2006), VIBRANT (Kieft et al., 2020), VirSorter (Roux et al., 2015), Virsorter2 (Guo et al., 2021), Phigaro (Starikova et al., 2020), PhageBoost (Sirén et al., 2021), DBSCAN-SWA (Gan et al., 2020), ProphET (Reis-Cunha et al., 2019), and Seeker (Auslander et al., 2020)—were each used to predict the prophages in 78 different manually curated microbial genomes.
Most of these programs utilize protein sequence similarity and HMM searches of core prophage genes to identify prophage regions. PhageBoost leverages a large range of protein features (such as dipeptide and tripeptide combinations) with a trained prediction model. PhiSpy was originally designed to identify prophage regions based upon seven distinct characteristics: protein length, transcript directionality, AT and GC skew, unique phage words, phage insertion points, optionally phage protein similarity and sequence similarity. DBSCAN-SWA likewise uses a range of gene metrics and trained prediction models to identify prophages. Seeker uses a new neural network model applied to bacterial and phage reference genome sequences to classify sequences as phage or bacteria. It was intended for use with sort sequences but can be used to identify prophages.
Regardless of whether gene annotations are available, Virsorter2, Phigaro, PhageBoost, and ProphET all perform de novo gene prediction with Prodigal (Hyatt et al., 2010) and VirSorter uses MetaGeneAnnotator (Noguchi et al., 2008) for the same purpose. VIBRANT can take proteins if they have ‘Prodigal format definition lines’ but otherwise performs predictions with Prodigal. PhageBoost can take existing gene annotations but this requires additional coding by the user. DBSCAN-SWA can take gene annotations or can perform gene predictions with Prokka (Seemann, 2014). PhiSpy takes an annotated genome in GenBank format and uses the gene annotations provided.
The prophage prediction packages Phigaro, PhiSpy, VIBRANT, VirSorter, and VirSorter2 are all able to be installed with conda from the Bioconda channel (Grüning et al., 2018), while Phispy, Phigaro, PhageBoost, and Seeker can be installed with pip—the Python package installer. Phigaro, VIBRANT, VirSorter, and VirSorter2 require a manual one-time setup to download their respective databases. Phigaro uses hard-coded file paths for its database installation, either to the user’s home directory or to a system directory requiring root permissions. Neither option is ideal as it is impossible to have isolated versions or installations of the program, and it prevents updating the installation paths of its dependencies. For PhageBoost to be able to take existing gene annotations, a custom script was created to skip the gene prediction stage and run the program. Basic PhiSpy functionality is provided without requiring third-party databases. However, if the HMM search option is invoked, a database of phage-like proteins—e.g. pVOG (Grazziotin et al., 2017), VOGdb (https://vogdb.org), or PHROGS (Terzian P et al., 2021)—must be manually downloaded before it can be included in PhiSpy predictions. DBSCAN-SWA is not currently available on any package manager and must be pulled from GitHub, however all its dependencies are available via conda and it could easily be added in the future. All the above “manual” installation and setup steps are uncomplicated and are automatically executed by the Snakemake pipelines provided in the framework.
Phage Finder was last updated in 2006 and is not available on any package manager that we are aware of. The installation process is dated with the package scripts liberally utilising hard-coded file paths. The Snakemake pipeline for this package resolves this with soft links between the framework’s directory to the user’s home directory (where the package expects to be installed). The dependencies are available via conda allowing the complete installation and setup to be handled automatically by Snakemake.
Installing and running ProphET is a non-trivial task. It requires the unsupported BLAST legacy and EMBOSS packages and a set of Perl libraries, including a custom library for preparing the necessary GFF files for running the program. The dependencies are mostly available via Conda and the remaining required files are included in this repository. LysoPhD does not appear to be available to download anywhere and was dropped from the comparison.
There are many potential ways to compare prophage predictions. For instance, is it more important to capture all prophage regions or minimise false positives? Is it more important to identify all the phage-encoded genes, or the exact locations of the attachment site core duplications (attL and attR)? We explore several metrics to highlight the different strengths of each prophage prediction tool. PhiSpy, VIBRANT, Phigaro, and ProphET performed best for mean accuracy (Figure 1a; Table 2) while Seeker and DBSCAN-SWA performed the worst. PhiSpy, Phigaro, Phage Finder, VIBRANT, and ProphET performed best for mean precision (Figure 1b; Table 2). Seeker, DBSCAN-SWA, PhageBoost, VirSorter, and VirSorter2 all performed poorly for mean precision. This was mostly driven by a high false-positive rate compared to the other tools (Figure S1). VirSorter, VirSorter2, VIBRANT, PhiSpy, DBSCAN-SWA, and PhageBoost all had high mean recall scores.
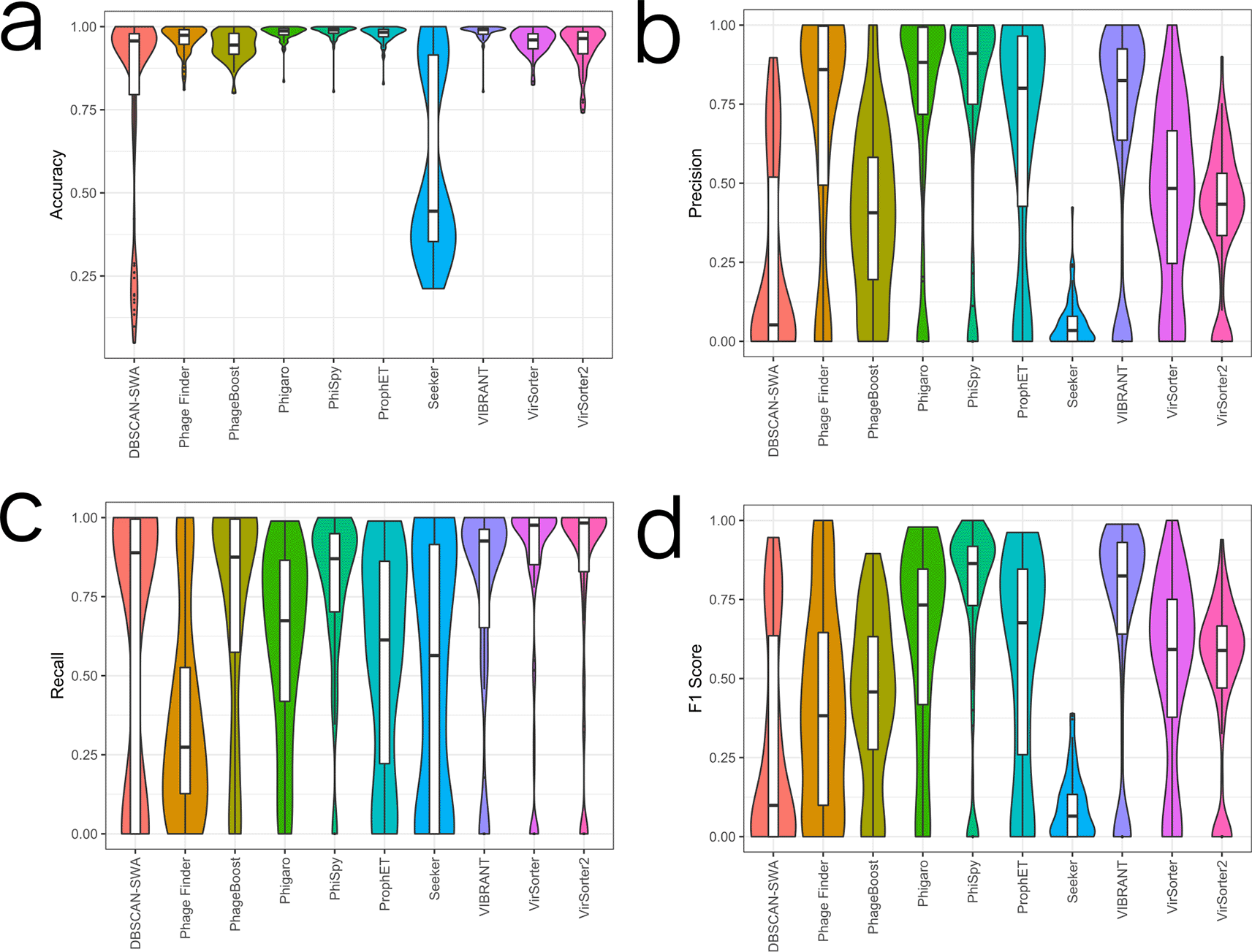
Violin plots for each tool are shown with individual points for each genome indicated. The graphs show: ‘Accuracy’ (a) as the ratio of correctly labelled genes to all genes, ‘Precision’ (b) as the ratio of correctly labelled phage genes to all predicted phage genes, ‘Recall’ (c) as the ratio of correctly labelled phage genes to all known phage genes, and ‘f1 Score’ (d) as defined in the methods. For all graphs, more is generally better.
Each tool balances between recall and precision. For example, the more conservative Phage Finder performed relatively well in terms of precision, making very confident predictions, but had one of the lower mean recall ratios and was not predicting prophages based on limited information. In contrast, the more speculative DBSCAN-SWA and PhageBoost both exhibited the opposite trend.
The f1 Score is a more nuanced metric, as it requires high performance in both precision and recall. PhiSpy, VIBRANT, Phigaro, ProphET, VirSorter, and VirSorter2 all averaged above 0.5, while the remaining tools suffered from many false predictions (FP or FN) (Figure 1d; Table 2).
Lastly, we visualised f1 scores for each genus across the tools to elucidate selection biases in the database (Figure S2a). Escherichia appeared to perform either very well or very poorly depending on the tool used but most genera appeared to be less variable between the different tools. We performed Mann-Whitney tests to compare the f1 scores of each genus against the other genera (Figure S2b). The f1 scores for Streptococcus, Staphylococcus, Listeria, and Burkholderia were significantly higher than the population average, and the f1 scores for, Ralstonia, Photorhabdus, Mycobacterium, Geobacter, Deinococcus, Cyanobacterium, Brucella, and Bifidobacterium were significantly lower than the average. An explanation for some of this variation would be that Streptococcus prophages for instance are well studied and highly conserved (Rezaei Javan et al., 2019), whilst other genera have been studied less, or their (pro) phages are highly diverse.
Many users will not be too concerned about runtime performance, for instance if they are performing a one-off analysis on a genome of interest all the tools will finish in a reasonable time. However, efficient resource utilization is an important consideration for large-scale analyses. Provisioning computing resources costs money and a well optimised tool that runs fast translates to real-world savings. The runtime distributions across the genomes are shown for each tool in Figure 2a and Table 3. The slowest prophage predictors were generally VirSorter and VirSorter2 with mean runtimes of 1,255 and 1,900 seconds respectively, except for a single DBSCAN-SWA run taking 5,718 seconds. Seeker was the fastest tool (6.65 seconds mean runtime), although this may not be a fair comparison given that multiple instances of this tool were not able to be run at the same time (see below). PhageBoost was the next fastest (37.8 seconds mean runtime) and Phage Finder, Phigaro, PhiSpy, and ProphET all performed similarly well in terms of runtime.
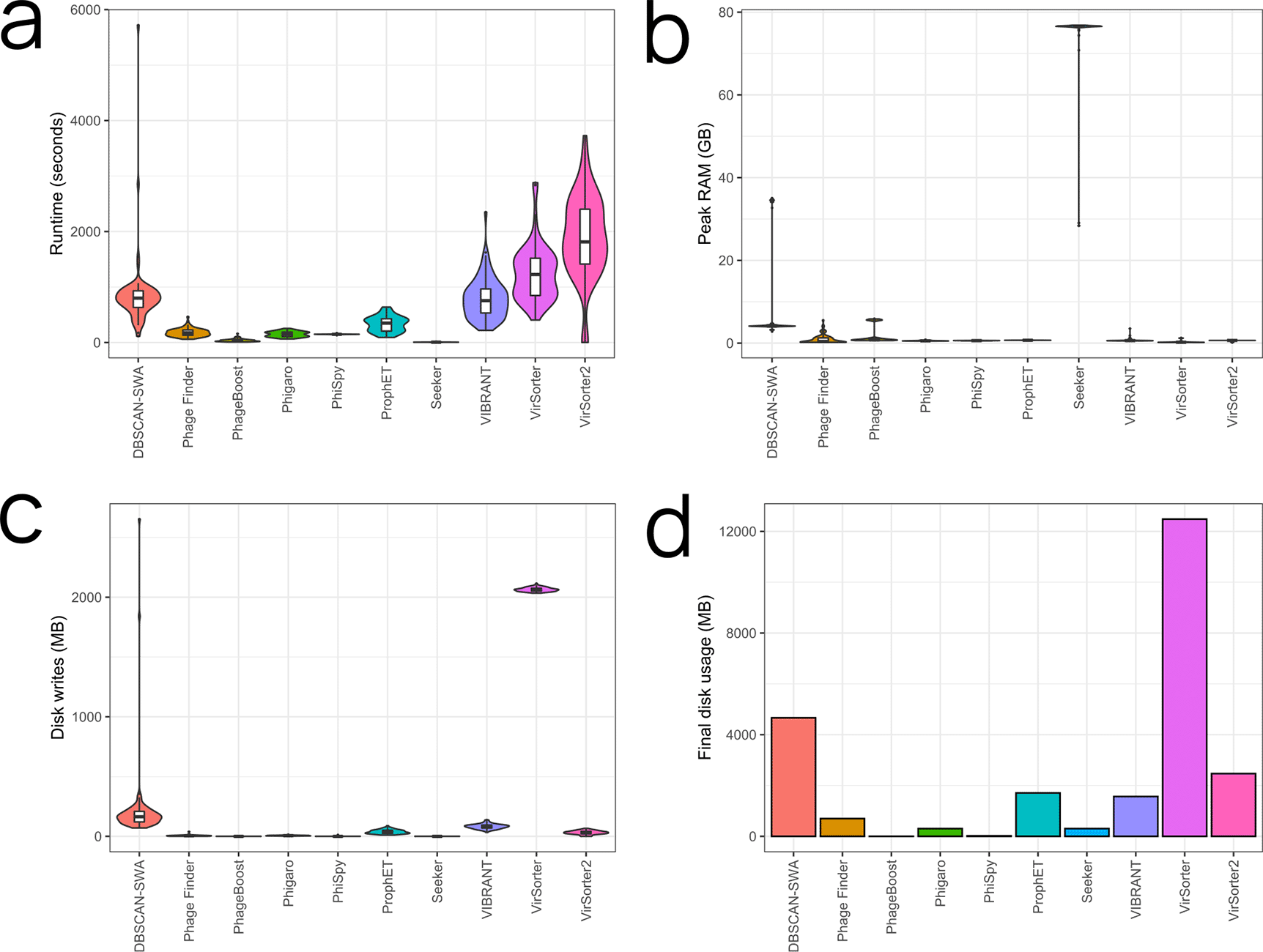
Violin plots for each tool are shown with individual points for each genome indicated. The graphs show total runtime in seconds (a), peak memory usage in MB (b), total file writes in MB (c) and the final total disk usage (all genomes) in MB (d). For all graphs, less is better.
Memory requirements also remain an important consideration for provisioning resources for large-scale analyses. For instance, inefficiency is encountered where the memory required by single-threaded processes exceeds the available memory per CPU. Peak memory usage for each tool is shown in Figure 2b and Table 3. Seeker had by far the highest mean peak memory usage at 75.2 GB. Our approach of running an instance of a tool on each system CPU failed for Seeker due to its extremely high peak RAM usage. We instead ran Seeker on a single CPU, one genome at a time. DBSCAN-SWA had the next highest mean peak memory of 8.76 GB. However, several runs required nearly 35 GB peak memory. Memory requirements were lowest for VirSorter with 287 MB mean peak memory, and mean peak memory usage for Phage Finder, Phigaro, PhiSpy, ProphET, VIBRANT, and Virsorter2 were all under 1 GB. Apart from Seeker and the DBSCAN-SWA outliers, there were no situations where the peak memory usage would prevent the analysis from completing on a modest personal computer. At larger-scales, Phage Finder, Phigaro, PhiSpy, ProphET, VIBRANT, VirSorter, and VirSorter2 have an advantage in terms of peak memory usage.
Another important consideration for large-scale analyses are the file sizes that are generated by the different tools. Large output file sizes can place considerable strain on storage capacities, and large numbers of read and write operations can severely impact the performance of a system or HPC cluster for all users. Total file writes for the default files (in MB, including temporary files) are shown in Figure 2c and the final disk usage for all genomes for each tool is shown in Figure 2d; these are also summarised in Table 3. VirSorter, DBSCAN-SWA, VIBRANT, ProphET, and VirSorter2 performed the most write operations. The other tools performed similarly well and have a clear advantage at scale as they perform far fewer disk writes. VirSorter and DBSCAN-SWA removed most of their generated files, however, the final disk usage for these tools were still the highest at 5.36 and 2.96 GB respectively. Disk usage for PhageBoost and PhiSpy was by far the lowest at 0.14 and 15 MB respectively.
Every bioinformatics comparison involves many biases. In this comparison, PhiSpy performs well, but we developed PhiSpy and many of the gold-standard genomes were extensively used during its development to optimize the algorithm. VirSorter and VirSorter2 were primarily developed to identify viral regions in metagenomes rather than prophages in bacterial genomes—although they have been used for that e.g. in Glickman et al. (2020)—and filtering VirSorter and VirSorter2 hits with CheckV (Nayfach et al., 2021) is recommended. Likewise, Seeker was designed for classifying short sequences as either phage or non-phage. It was trained on phages, not prophages, and was not originally intended for classifying prophage regions in complete genomes. Furthermore, the current database of prophage annotations is heavily skewed towards more heavily studied phyla, and we show that there can be significant differences in performance depending on taxonomy. By openly providing the Prophage Prediction Comparison framework, creating a framework to install and test different software, and defining a straightforward approach to labelling prophages in GenBank files, we hope to expand our gold-standard set of genomes and mitigate many of our biases. We welcome the addition of other genomes (especially from beyond the Proteobacteria/Bacteroidetes/Firmicutes that are overrepresented in our gold-standard database).
Recent developments in alternative approaches to predict prophages, including mining phage-like genes from metagenomes and then mapping them to complete genomes (Nayfach et al., 2021) and using short-read mapping to predict prophage regions from complete bacterial genomes (Kieft & Anantharaman, 2021) have the potential to generate many more ground-truth prophage observations. However, both approaches are limited, as they identify prophages that are active, but not quiescent prophage regions. Thus, they will provide useful true positive datasets for prophage prediction algorithms but may not provide accurate true negative datasets.
Establishing a gold-standard dataset of prophage and associated metrics is critical to enable a robust comparison of prophage prediction tools. In particular, the current comparison suggests that most tools perform reasonably well by themselves to detect phage-like regions in complete bacterial genomes, and most of the differences between tools stem from different trade-offs between precision and recall with default parameters and different compute resource requirements. Specifically, using the gold-standard dataset and the metrics defined here, PhiSpy, VIBRANT, and Phigaro were the best performing prophage prediction tools for f1 score. Phage Finder performs well in terms of precision at the expense of false-negatives, whereas VirSorter, VirSorter2, DBSCAN-SWA and PhageBoost perform better for recall at the expense of false-positives. Currently, Seeker, DBSCAN-SWA, VirSorter, and to a lesser extent VirSorter2 are not as well suited for large-scale identification of prophages from complete bacterial genomes when compared to the other tools, and would require using custom cutoffs and/or post-processing predictions with another tool such as CheckV. In terms of runtime performance, PhiSpy and Phigaro were among the best in every metric, and ProphET and VIBRANT performed well in most metrics. These comparisons are, however, relying on expert curation of a limited number of genomes. More genomes with manually curated prophage annotations are thus needed as well as a larger number and diversity of experimentally-verified prophages, and we anticipate that these benchmarks will change with the addition of new genomes, the addition of new tools, and as the tools are updated over time. We intentionally designed the current framework to be easily amendable and expanded, and developers are strongly encouraged to contribute by adding or updating their tool and adding their manually curated and/or experimentally verified genomes to be included in the benchmarking. Users are strongly encouraged to check the GitHub repository for the latest results before making any decisions on which prophage prediction tool would best suit their needs.
RAE conceived of the study; KM and PD generated the initial gold-standard set and MJR, SKG, LI, and EP contributed to the current gold-standard set; RAE and MJR created the framework; RAE, MJR, SR, MM, and JB performed the analysis. All authors contributed to the manuscript writing.
This work supported by the National Institute Of Diabetes And Digestive And Kidney Diseases of the National Institutes of Health under Award Number RC2DK116713 to RAE. The support provided by Flinders University for HPC research resources is acknowledged. The work conducted by the U.S. Department of Energy Joint Genome Institute (SR), a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.
Zenodo: linsalrob/ProphagePredictionComparisons: Review release. https://doi.org/10.5281/zenodo.4739878. (Roach & Edwards, 2021b).
This project contains the following underlying data;
Underlying data and the prophage annotated GenBank files are also available at GitHub: Comparisons of multiple different prophage predictions https://github.com/linsalrob/ProphagePredictionComparisons/tree/v0.1-beta (Roach & Edwards, 2021a). Please note that you will need Git (git-scm.com) and Git LFS (git-lfs.github.com) to retrieve the GenBank files from the GitHub repository. Support for these files and framework pipelines are available via GitHub issues.
Zenodo: Extended data for ‘Philympics 2021: Prophage Predictions Perplex Programs’: https://doi.org/10.5281/zenodo.4739878.
This project contains the following extended data:
SupplementaryTables.xlsx:
FigureS1.png:
• Figure S1. False positive comparison. Violin plots for each tool show ‘False Positives’ as the number of genes incorrectly labelled prophage genes in each genome. Less is better.
FigureS2.png:
• Figure S2a. F1 scores by genus for each tool. F1 scores are shown for each genome grouped and coloured by genus and separated by prophage caller.
• Figure S2b. F1 score distribution for each genus for all tools. Boxplots of F1 scores for each genome over all tools, grouped and coloured by genus. P-values and significance are indicated for Mann-Whitney tests that were performed to determine if a genus had significantly higher or lower F1 scores when compared to the other genera.
prophageAnnotation.md
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)