Keywords
SARS related coronavirus, SARS-CoV-2, SARS-CoV-1, COVID-19, virus artificial synthesis, yeast S. cerevisiae, directed evolution, genomic transformation, genome editing, synthetic biology
This article is included in the Cell & Molecular Biology gateway.
This article is included in the Coronavirus (COVID-19) collection.
SARS related coronavirus, SARS-CoV-2, SARS-CoV-1, COVID-19, virus artificial synthesis, yeast S. cerevisiae, directed evolution, genomic transformation, genome editing, synthetic biology
See the author's detailed response to the review by Alexander Y Panchin
See the author's detailed response to the review by Federico Di Lello
28th April 2025: As agreed with the author(s), the peer review for this article has been discontinued. This means that the article is no longer under peer review at F1000Research, and is not indexed in PubMed, Scopus and other bibliographic databases.
From the beginning of the COVID-19 pandemic, in March 2020, evidence was put forward that the outbreak of novel coronavirus SARS-CoV-2 within the human population was most likely a product of natural evolution1. According to this view, COVID-19 is a zoonosis that probably originated from a species of closely related bat coronaviruses2. Prior to a hypothetical spillover event, a recent ancestor to SARS-CoV-2 likely evolved inside bat host cells for many decades3. However, the natural evolution hypothesis of SARS-CoV-2 origin is currently not without considerable limitations: first, the difficulty in characterizing the evolutionary origin of the unusual poly-basic (PRRAR) furin cleavage site (FCS) at the S1/S2 junction of the SARS-COV-2 spike (S) glycoprotein4; second, the discrepancy between an exponentially suppressed tropism of SARS-CoV-2 in Rhinolophus sinicus bat cells5 and the high susceptibility of SARS-CoV-2 toward cell entry via Rhinolophus sinicus angiotensin-converting enzyme 2, its primary entry receptor6; and third, the persistent inability to identify an intermediate ancestral host between human and the horseshoe bat Rhinolophus affinis. This species was reported to be the host of coronavirus RaTG137,8, currently the isolate with the closest evolutionary relationship to the SARS-CoV-2 genome9, which is located on the same phylogenetic branch as Rhinolophus sinicus bat coronavirus10. Finding the last animal progenitor host of SARS-CoV-2 has been further complicated by the fact that RaTG13 lacks a homologous FCS sequence, and by a continued uncertainty about the origin of RaTG13 itself11,12. Thus even in the third year after the emergence of COVID-19, a more closely related evolutionary progenitor sharing naturally the unusual functional characteristics, like the S1/S2 FCS, with SARS-CoV-2 has yet to be found in China13,14, or outside15.
In contrast to the natural evolution hypothesis for SARS-CoV-2, the above limitations do not necessarily apply to genetic engineering of viral genomes in laboratory environments. For example, the theory that SARS-CoV-2 could be the product of laboratory manipulation involving a passage through cell culture has been critically discussed1. In addition, for SARS coronavirus, it has long been established that introducing a synthetic poly-arginine construct at the furin cleavage site significantly increases the rate of entry into human cells compared with wild-type spike protein16. Also before 2010, after a period of rapid progress in the understanding the relevant host-virus factors17,18, natural barriers in host range of RNA viruses were rationally extended, leading to artificial genome assembly and directed viral replication in new species including model organisms that originally were not permissive, such as the yeast Saccharomyces cerevisiae19,20. Accordingly, to transform budding yeast into a artificial host for viral synthesis and replication, the general scheme for both positive and negative sense RNA has been to co-express a viral RNA dependent RNA polymerase (RdRp) and, if also necessary for replication, additional factors on plasmids under the control of auxotrophic yeast selectable markers (YSM)20,21. For betacoronaviruses, the genus to which all SARS-like coronaviruses belong, the key experimental step had been described already in 2002 by Yount et al., where the essential genomic replicase domain located between nsp3 (non-structural protein 3) and RdRp was cloned and robustly expressed in yeast from a standard pYES2 vector carrying the URA3 (uracil requiring orotidine-5'-phosphate decarboxylase) gene as its only auxotrophic marker22. Yeast selectable (auxotrophic) markers, and specifically URA3, have been since described and used experimentally to direct cell lines into stable expression of a large variety of virus derived cDNA constructs and clones, including many human pathogens such as recombinant SARS coronavirus (see columns 14–15 in 20). At the same time, plasmids with YSMs had already been known to function as entry gates for directed insertion of exogenous genetic material into yeast chromosomes23. This insertion process, by means of homologous recombination, is a priori independent of both transcription and the optional RdRp driven RNA replication cycle. Following this rationale, Thao et al.24 have more recently demonstrated that, prior to transcription into infectious RNA, several overlapping genomic domains efficiently assemble into a full-length SARS-CoV-2 coronavirus clone on a yeast artificial chromosome (YAC) using HIS3 (histidine requiring imidazoleglycerol-phosphate dehydratase) as YSM. YAC assembly therefore facilitates recombination with endogenous host chromosomes resulting in viral RNA or infectious clones with “traces of yeast genomic DNA”24. Our hypothesis is that such artificial synthesis in yeast cells would leave behind traces in the genomic sequences of both the virus construct and the synthetic host.
SARS and SARS-like betacoronavirus whole genome nucleotide sequences were selected following the comprehensive sequence and phylogenetic analyses by Zhou et al.25 and from Li et al.10. In our study, sequences were selected only if they had a valid GenBank accession identifier or an NCBI Reference Sequence (RefSeq) accession identifier, as of 5 June 2021, resulting in the reference set of 13 whole genome virus sequences (see also Extended Data). This set was extended by 5 additional genomic sequences, BANAL 20-52/20-103/20-236, icSARS-CoV-2, and rSARS-CoV-2 YAC (see, Repository-hosted data, for the full list). BLAT whole genome comparative sequence analysis was performed using the BLAT public webserver (BLAT, RRID:SCR_011919) with options set “Genome: Search all” and “All results (no minimum matches)”. Each BLAT search from the above set of query sequences against the entire multi-species genome database resulted in a high number of tiles, i.e. perfectly aligned short DNA sequences of length 11, to the yeast S. cerevisiae (SacCer3/S288c). BLAT identified many homologous regions by aggregating multiple tiles, and to each homologous region it produced an integer score S, which is the number of perfectly matched positions therein. Each of the 18 corresponding BLAT genomic alignments to the yeast S. cerevisiae (Extended data Tables S2 – S19) produced a profiled BLAT score, pS, which was the genome-wide distribution of S scores (output table column [SCORE]) weighted by the corresponding length of the homologous genomic region (output table distance between columns [START] and [END]). To remove its shortest-scale fluctuations, these profiles were smoothed by a centered sliding window filter with window size of 200 nucleotides (nt). The cumulative profiled BLAT score, cS, was the total sum over this distribution (excluding matches to mitochondrial DNA). Using cS, a genome-wide measure of yeast homology was generated through the statistical null hypothesis that those profiles, for which no BLAT yeast peaks with pS > 20 were detected, followed a normal distribution N(0,1) in their standardized cS values. This distribution was therefore sampled by shifting cS values by the sample’s mean and dividing by its standard deviation. The resulting standardized BLAT p-values, returned by the normal cumulative distribution function and transformed into negative logarithms, became a statistical test of the above null hypothesis and, as such, a measure of sequence homology with S. cerevisiae. A statistical significance (chosen above a level of 0.05) test for pairs of p-values, p1 < p2 , was calculated with conditional probabilities p1/p2. Negative log p-value for rSARS-CoV-2 YAC was the average of 12 Sanger–sequenced yeast artificial chromosomes with detected mutations (relative to SARS-CoV-2 Wuhan-Hu-1 reference genome) mapped onto the synthetic genome construct rSARS-CoV-2 with sequence deposited at Genbank MT108784, see Extended Data Table 4 in Thao et al.24. Sequence alignments for cross-validation were produced with LALIGN from the fasta36-36.3.8/bin/lalign36 software package (version number 36.3.8) with parameter settings: -f -12 -g 0 -E 1. This parameter choice followed standard parameters for LALIGN. Sequence identities were calculated using the Clustal Omega public webserver (RRID:SCR_001591) with standard preset parameters. Nucleotide sequence database searches were performed with the NCBI blastn webserver (RRID:SCR_001598) against the entire ‘Nucleotide collection (nr/nt)’ restricted to eukaryotic (taxid:2759) ‘genomic DNA’ sequence records deposited before the year 2020. The reason behind leaving out sequencing data generated after 2019 is growing evidence, since the beginning of the COVID19 pandemic, of exogenous genomic integration in cultured cells and in the infected host26,27, as well as widespread contamination of laboratory environments with SARS-CoV-2 cDNA28; this dissemination of SARS-CoV-2 cDNA into the host’s natural environment may cause sequencing-based virus testing anomalies28, and has already resulted in chimeric virus-host sequences in reference databases unseen before 2020 (e.g., https://www.ncbi.nlm.nih.gov/bioproject/PRJNA720932). Therefore, by restricting searches to records before 2020, the likelihood of assigning such false positive sequence hits to the pre-pandemic origins of SARS-CoV-2 would be minimized in our study. Also, ‘Models (XM/XP)’, partial, and predicted sequences were excluded. blastn algorithm parameters were set at standard values except for E-value threshold (100 instead of 0.05), and gap cost (6 instead of 5).
To interrogate the possibility that a similar passage through yeast cells took place within the family of SARS coronaviruses, we initially selected eight reference genomes25 for further analysis (see Methods): SARS-CoV-2 isolate Wuhan-Hu-1 (GenBank reference NC_045512.2), Rhinolophus affinis bat coronavirus RaTG13 (MN996532.2), Rhinolophus pusillus SL-CoV ZXC21 (MG772934.1), Rhinolophus pusillus SL-CoV ZC45 (MG772933.1), Rhinolophus acuminatus bat coronavirus RacCS203 (MW251308.1), Rhinolophus cornutus bat coronavirus Rc-o319 (LC556375.1), SARS-CoV Urbani (AY278741.1), and MERS-CoV isolate HCoV-EMC/2012 (NC_019843.3). For comparative genomic sequence analysis we used a standard bioinformatics approach with the BLAST-like Alignment Tool (BLAT) (BLAT, RRID:SCR_011919)29. The rationale was that BLAT, a more accurate genome sequence alignment tool than other conventional approaches29, would detect such traces of yeast DNA. In line with this hypothesis, a large majority of BLAT matches was on the same two target genomes (see also Extended data Table S1): SARS-CoV-2 (NC_045512.2), a self-match to the only lineage b betacoronavirus genomic sequence in the BLAT database, and S. cerevisiae (SacCer3/S288c). To obtain a genome-wide view of this yeast homology pattern we stacked together all homologous regions weighted by their individual alignment scores S, which resulted in an accumulated homology profile, pS (see Methods and Extended Data Figures S1 and S2).
For SARS-CoV-2, two prominent (pS > 20) peaks indicated highly localized profile scores at levels ~10-fold above the apparent background. A first peak (P1) reaching a top alignment score of 47 in the narrow genomic interval [7191..7192]max, and a second peak P2 over ~18,000 bases downstream with a score of 36 in the region [25196..25212]max (see, Figure 1). To put these data into an established gene-function context these two maxima, with half-maximum widths w1/2 = 215 and w1/2 = 219, respectively, were annotated with available information from the closest and most specifically annotated genomic region in RefSeq, the NCBI Reference Sequence database30. Thus P1 was closest to the start of the C-terminal domain of non-structural protein 3 (designated nsp3C), which extends over the interval [6962..8552]. The C-terminal domain of nsp3 is known to play a critical role in replication due to its direct interaction with nsp4, thereby facilitating virus-induced membrane rearrangement and replication complex formation; conversely, loss of nsp3C-nsp4 interaction abolishes SARS coronavirus replication31. P2 was located toward the 3′ end of the open reading frame of the spike gene. Here it overlapped with the 3′ end of the stretch that covers both the S1/S2 cleavage region and the S2 fusion subunit of the S protein (S_S1/S2, with interval [23192..25187]). The S_S1/S2 domain includes the characteristic furin cleavage site at the S1/S2 junction32, which has previously been described as unique to SARS-CoV-2 among lineage b betacoronaviruses4. Cleavage activates the nearby S2 fusion peptide and together they constitute an essential part in SARS-CoV-2 particle-dependent and particle-independent cell entry through fusion of viral and cellular membranes33,34. A similar analysis for the RaTG13 viral genome identified only one isolated peak (P3) with a maximum profile score of 50 on the interval [9713..9733]max, and with w1/2 = 230. It intersected with the coding region of the C-terminal domain of nsp4 located at [9770..10046] (Figure 1).
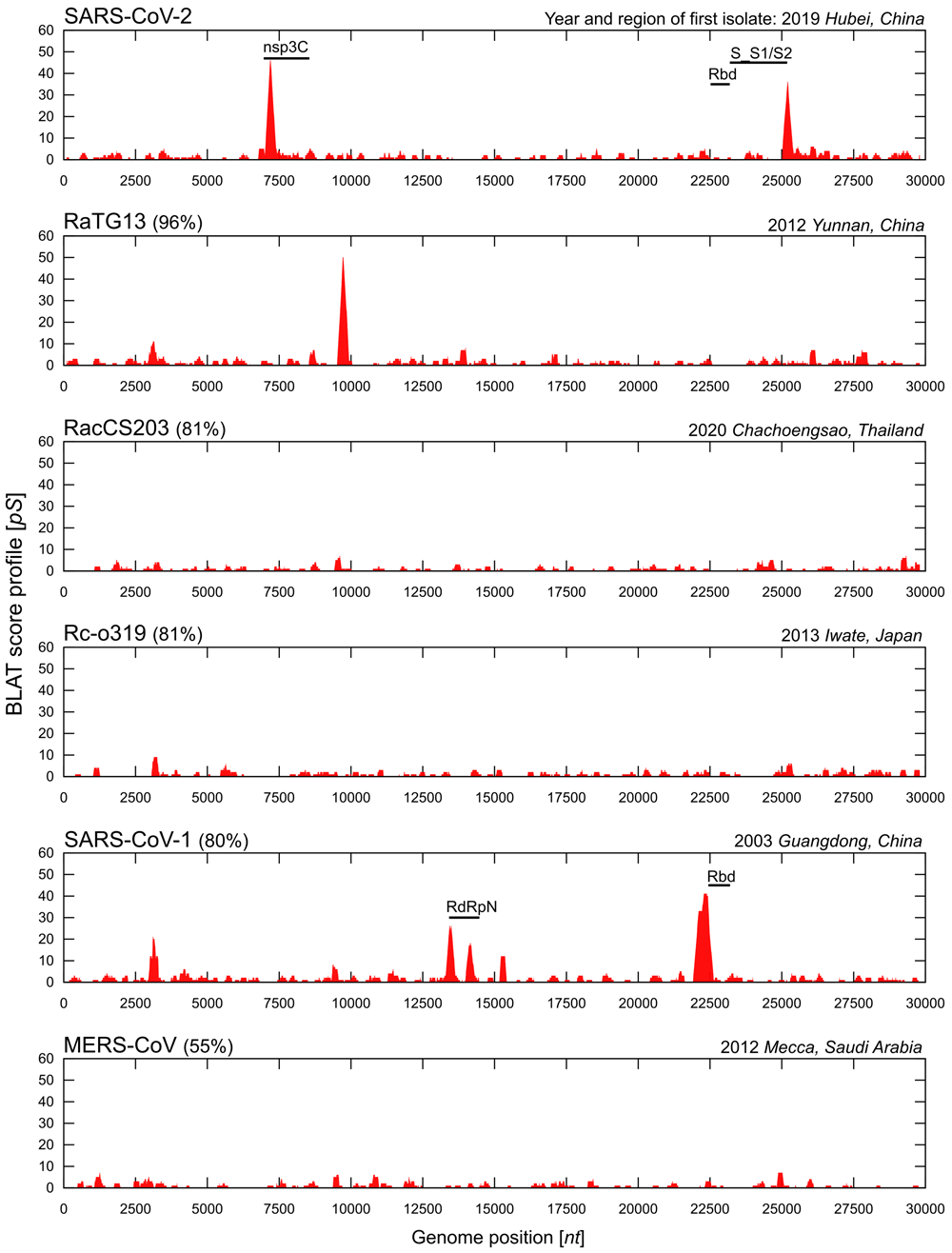
Alignment scores from hits matching S. cerevisiae full genomic sequence assembly SacCer3/S288c. For the corresponding BLAT output, see Table S1, and Table S2–S9. Upper left, in brackets, percent sequence identity of query genome to SARS-CoV-2. Profiles are ordered by decreasing sequence identity to SARS-CoV-2. Of note, detected yeast homology patterns, nucleotide sequence similarity, and geographic location (region, country) do not converge. nsp3C, non-structural protein 3 C-terminal domain [YP_009724389.1 (2,232..2,762)]; Rbd, receptor binding domain [SARS-CoV-2: YP_009724390.1 (319..541); SARS-CoV-1: AAP13441.1 (317..569)]; S_S1/S2, spike (S) protein S1/S2 domain cleavage region and the S2 fusion subunit [YP_009724390.1 (543..1,208)]; RdRpN, N-terminal region of the RNA dependent RNA polymerase [AAP13442.1 (4,383..4,735)].
Of special interest in this analysis was a 16 base sequence (TTCTCCTCGGCGGGCA) near P2 between position 23599 and 23614, which corresponded to the furin cleavage site and identically aligned with bases [810386..810401] from S. cerevisiae chromosome XIII. In the forward +1 reading frame this sequence encodes the amino acids SPRRA and thus includes the critical PRRA insert in SARS-CoV-2. This shared sequence could be extended to 17 consecutive nucleotides (TTCTCCTCGGCGGGCAA), which are identically found in known SARS-CoV-2 variants that emerged after serial passage in cell culture (e.g., GenBank entry MZ995185.1), and—at codon level— are also compatible with the entire ancestral SPRRAR motif. As such, TTCTCCTCGGCGGGCAA represented the longest identical nucleotide sequence between SARS-CoV-2 clade and S. cerevisiae lineage that covered the furin cleavage site. To test the specificity of TTCTCCTCGGCGGGCAA across potential eukaryotic host organisms, we performed BLAT and standard blastn sequence searches. For BLAT, no hits were found except for the one in yeast. When restricted to ‘genomic DNA’ sequence records dated before 2020, an extensive blastn search among all GenBank eukaryotic genomic sequences produced no identical sequence hits other than the Saccharomyces cerevisiae match above (see, Extended Data File S1). A similar result was obtained when potential host specificity was tested with the shorter TTCTCCTCGGCGGGCA sequence (Extended Data File S2 and S3), as well as with the entire SARS-CoV-2 genomic sequence (Extended Data File S4 and S5). These data specifically identified the yeast S. cerevisiae as a potential genomic recombination donor of the critical FCS in the spike protein of SARS-CoV-2.
In the SARS coronavirus Urbani genome (SARS-CoV-1), two additional signals were detected: P4 with a maximum score pS = 26 at position [13486..13497]max and w1/2 = 222; and a broader second peak, P5, with pS = 41 at position [22286..22391]max and w1/2 = 477. P4 sharply co-localized with the N-terminus of the RdRp domain at [13414..14470]. P5 was annotated with the N-terminal part of the spike gene’s receptor binding domain (Rbd) located in the interval [22443..23199]. In contrast to the five signals identified in these three genomes, an equivalent analysis for the other five (RacCS203, SL-ZC45, SL-ZCX21, Rc-o319, MERS-CoV) produced only negative results. Their accumulated homology profiles were evenly distributed across the entire genomes consistent with a low random score background from many short spurious matches. As a further specificity control, negative results were obtained (see, Figure S3 and Tables S10–S14) after profiling the five most closely SARS-CoV-1 related betacoronavirus isolates from five wild animals (civet, Paradoxurus hermaphroditus, Paguma larvata, Aselliscus stoliczkanus, and Rhinolophus sinicus), which together with SARS-CoV-2 occupy the same phylogenetic branch10. These data collectively produced a differential yeast homology signature, with only SARS-CoV-1, SARS-CoV-2 and RaTG13 statistically significant, after calculating standardized p-values (Figure 2) from the entire BLAT profiles to all 13 of the above sequences (Tables S2–S14). This analysis also included the three recently identified bat SARS-like coronavirus genomic sequences from the same clade as RaTG13, i.e., BANAL-20-52, BANAL-20-103, and BANAL-20-236 (Tables S15-S17), none of which yielded statistically significant p-values. To cross-validate the detected yeast homology signals in P1- P5, we also used an independent sequence alignment method, LALIGN35, which additionally produced statistics (E-values) for pairwise alignments. While the peaks P1 and P2, as well as P4 and P5, could be positively validated, the P3 signal in RaTG13 detected by BLAT did not yield a statistically significant alignment with LALIGN, with its E-value reaching above 0.01 (see, Table S21 and Figure S4). Taken together, these highly differential data show that, for SARS-CoV-1 and for SARS-CoV-2, genes known to be critical for viral replication and host cell invasion display localized yeast homology at their flanking regions with limited extensions into the corresponding open reading frames.
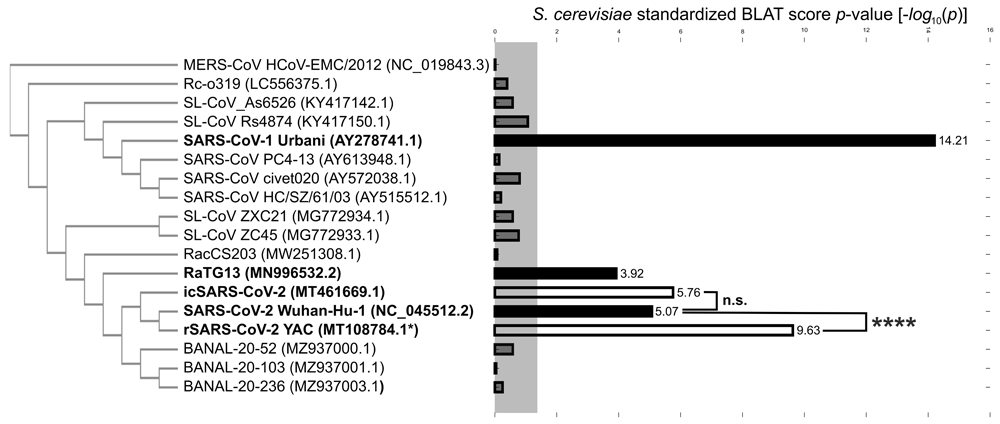
Individual p-values were calculated from sampled means and standard deviations in BLAT outputs (see, Table S2–S19 and Methods). Grey shaded box depicts 0.05 significance level. Pairwise statistical significance test by conditional p-values (see, Methods); n.s., not significant. The negative log p-value for rSARS-CoV-2 YAC (MT108784.1*) was the average over 12 such values from sequenced YAC clones, see Thao et al. 2020 and Methods. Evolutionary guide tree (cladogram) generated by sequence identities between full genomic sequences (see, Table S20).
As a further validation of our method, we turned to two derived genomic sequences of SARS-CoV-2: the recombinant rSARS-CoV-2, assembled through reverse genetics into a YAC24, and the infectious clone icSARS-CoV-2, assembled without yeast through a cell-free in vitro ligation method36. Even though their genomic sequences were identical to SARS-CoV-2 Wuhan-Hu-1 at >99.9% level (see, Table S20), our data (Figure 2) differentiated between rSARS-CoV-2 YAC, which relative to SARS-CoV-2 Wuhan-Hu-1 yielded a significant (p<0.0001) increase, and icSARS-CoV-2, which produced no significant difference to this SARS-CoV-2 reference genome. These data suggest that our approach may sensitively and specifically detect traces from a given yeast artificial synthesis history in recombinant SARS-CoV-2.
To explain the observed yeast DNA enrichment pattern in SARS coronavirus genomic sequences, we propose the following artificial synthesis model (Figure 3A): Its starting point is a doubly auxotrophic, synthetic yeast cell line with stable, heterologous expression of a viral replicase complex (RdRp, optionally together with auxiliary factors for replication, Aux) from a plasmid under the control of a selectable marker YSM1. A second plasmid carries another auxotrophic yeast selectable marker YSM2, which originates from a different chromosome, and regulates the expression of a non-replicative segment encoding for viral RNA (nrvRNA1). At this point, nrvRNA1 is any uninterrupted DNA segment from a SARS coronavirus related genome. Through homologous recombination, the target yeast chromosome is transformed and nrvRNA1 is integrated23 at the chromosomal site of the auxotrophy conferring allele homologous to YSM2. During cell growth double stranded DNA breaks occur, and breaks at both ends of nrvRNA1 ends, their flanking regions, and their homologous extensions into YSM2 are repaired preferably by intra-chromosomal gene conversion37, i.e. through a non-crossover homologous recombination, and with the endogenous site as the homologous repair donor (Figure 3A).
(A) First stage assembly and transformation in the artificial host S. cerevisiae of a plasmid encoded, non-replicable viral RNA (nrvRNA1) originating from a SARS-CoV related virus. Primary integration of non-homologous nrvRNA1 sequence occurs through homologous recombination (HR) between the auxotrophic plasmid yeast selectable marker YSM1 (grey box) and its chromosomal homolog (striped grey box); higher-order homologous recombination follows on the flanking regions of nrvRNA1 through intra-chromosomal gene-conversion; co-expression of viral replicase complex (RdRp) and other auxiliary viral genes (Aux). Scheme in parts adapted from Compton et al. (1982), and from Alves-Rodrigues et al. (2006). P, yeast promoter; An, poly-adenosine sequence. (B) Integrated profile scores, cS, from BLAT sequence hits on S. cerevisiae by chromosome number from the same six input sequences as in Figure 1 (purple columns); cS, score profile sum with cutoff pS > 30. Without a cut-off (pS > 0), the same order emerged (black horizontal bars, maximum pS score at each chromosome; all other maximum pS scores from the other genomic queries are below, within shaded area). Five common yeast selectable markers are assigned to their chromosomes of origin. (C) Inferred second stage for the synthetic biogenesis of SARS-CoV-2 and SARS-CoV-1. Yeast selectable markers pairings (YSM1, YSM2) matched in (B), chromosomal transformation by three segments nrvRNA1, 2, 3 transcribes into a virus (+)sense RNA, while also recombining with a given yeast artificial chromosome (YAC). Virus-like particle (self-)assembly follows by expression of the structural proteins S, E, M, and N from an enhanced plasmid set Aux*. Rz, self-cleaving ribozyme; YC, yeast chromosome.
If we assume that nrvRNA1 itself contains sequences homologous to the YSM1 carrying plasmids, e.g. through ends with overlaps, then the above model implies that higher-order integration events23 will occur between the YSM1 plasmid and the primary site of integration. In effect, short segments from its YSM1 region will be also integrated into nrvRNA1. In this case the model specifically predicts that during S. cerevisiae growth nrvRNA1 will accumulate sequences from two yeast chromosomes, i.e. those two which YSM1 and YSM2 originated from.
To test this prediction, we produced the score profile pS, but this time from the yeast sequence hits on each chromosome. For direct comparison, we then transformed each profile into a single number (cS), for all 16 chromosomes (mitochondrial chromosome excluded), by calculating the sum of pS over the entire chromosome length conditional on the cutoff pS > 30. In the case of SARS-CoV-2, this procedure resulted in two distinct peaks at chromosome number II and number XV (Figure 3B). For SARS-CoV-1, the highest two peaks were at chromosomes IV and V, followed by a much shallower peak on XVI with only 0.24 the height of IV. One peak was detected for RaTG13, also at XVI, whereas the other viral genomes produced no signal at the chosen cutoff (see, Figure 3B, also for similar data without a cutoff). To further connect these data to our model, we attempted to match the seven most commonly used auxotrophic yeast selectable markers38,39 according to their chromosomal origin: ADE2 (adenine requiring phosphoribosylaminoimidazole carboxylase, on chromosome XV), HIS3 (histidine requiring imidazoleglycerol-phosphate dehydratase, chr. XV), LEU2 (leucine requiring Beta-isopropylmalate dehydrogenase, chr. III), LYS2 (lysine requiring aminoadipate reductase, chr. II), MET15 (methionine requiring O-acetyl homoserine-O-acetyl serine sulfhydrylase, chr. XII), URA3 (uracil requiring orotidine-5'-phosphate (OMP) decarboxylase, chr.V), and TRP1 (tryptophan requiring phosphoribosylanthranilate isomerase, chr. IV). In agreement with the model prediction, five out the seven markers could be matched to the four highest of the five chromosome peaks detected in SARS-CoV-2 and SARS-CoV-1 (Figure 3B). For SARS-CoV-1 there was a marked URA3 associated peak (on chromosome V) with a yeast score that exceeded all other observed values by at least 2 orders of magnitude. For SARS-CoV-2, the maximum peak was associated with HIS3 (and ADE2) selectable markers (on chromosome XV). These data imply that for SARS-CoV-2 the two auxotrophic markers (YSM1, YSM2) could be any pair from the triple (HIS3, ADE2, LYS2), and for SARS-CoV-1 the pair (URA3, TRP1). Thus SARS-CoV-1 and SARS-CoV-2 both did, but RaTG13 did not completely fit into this artificial yeast model.
These results allowed us to infer a scheme for the artificial biogenesis of SARS-CoV-2 and SARS-CoV-1 in transformed yeast cells (Figure 3C). A minimum of three genomic fragments, designed through reverse genetics to assemble into a YAC, provide two outer DNA clone complements of a chosen progenitor SARS viral genome together with the inner segment nrvRNA1. For transformation, integration and assembly, the plasmids carry a YSM2 selectable marker with either the 5′-end (nrvRNA2) or the 3′-end (nrvRNA3) of the target virus genome, each with a specific overlap into both nrvRNA1 ends (regions 1′ and 1′′, respectively, see Figure 3C). Essential plasmid ingredients are also a transcriptional promoter for nrvRNA2, and a self-cleaving ribozyme (Rz) sequence for the correct 3′-end in nrvRNA319. Once these virus genomic RNA encoding segments are integrated into a yeast endogenous chromosome, homologous recombination with the YAC (if concurrently present) and genomic transcription of viral RNA follow. In contrast to the targeted sequence of the YAC, which was designed to not express yeast DNA, the recombinant viral DNA from the transformed chromosome is homologous to the entire YAC while also enriched with yeast genomic DNA. Virus RNA replication then commences upon its further transfection into replication competent host cells, or through additional co-expression of a viral replicase complex (RdRp and Aux, controlled through the auxotrophic marker YSM1, Figure 3C). A final optional step, assembly into a viral particle, may be achieved with a yeast virus-like-particle (VLP) expression system for the structural proteins S, E (envelope), M (membrane), and N (nucleocapsid) that can be expressed from an auxiliary plasmid, Aux*40.
Our results reveal a previously unidentified, highly differential sequence pattern in SARS-CoV-2 and SARS-CoV-1 genomes, which—according to our model—points to their history of targeted transformation, integration and recombination in an artificial S. cerevisiae host. This orthogonal layer of genomic sequence information significantly deviates from the standard reconstructed natural evolutionary history of lineage b (Sarbecovirus) coronaviruses by indicating a yeast artificial origin of SARS-CoV-1 and SARS-CoV-2. At the same time, our data robustly excludes all other analyzed clade members from this type of yeast artificial origin. A special case is RaTG13, which in our analysis produced both a simpler pattern and a weaker signal of common genetic history with yeast than the two mutually more similar homology signals found in SARS-CoV-1 and SARS-CoV-2. Yet RaTG13 is claimed to be much closer to SARS-CoV-2 evolutionarily7, i.e. 96% genomic sequence identity to SARS-CoV-2 against 80% between SARS-CoV-1 and the latter. This divergence suggests that if RaTG13 is assumed to be a product of natural evolution then both the sequences of SARS-CoV-1 and SARS-CoV-2 cannot be. Alternatively, the origin of RaTG13 could be artificial12 —along with SARS-CoV-2 and SARS-CoV-141, as our results also suggest. As a controversial candidate for a natural ancestral or intermediate SARS coronavirus host, palm civets had in fact never been identified as the original animal reservoir of SARS coronavirus, and a conclusive zoonotic host identification or characterization of a natural origin has not been given either. For example, the frequently cited work by Kan et al. concluded that “when SARS-CoV-like virus arrives at an animal market, the majority of palm civets, if not all, will become infected, and that the virus will evolve rapidly in animals to cause disease. Therefore, it is critical to identify the original animal reservoir to remove the continuing threat of SARS.“42 This conclusion, and further evidence that palm civets were not even an intermediate host, were supported by phylogenetic analysis for the initial stages of the SARS epidemic43, where a rooted phylogenetic tree placed the earliest human virus lineage before the first civet infections, and with both viral lineages originating from an unknown reservoir in late 2002. To date, this uncertainty and controversy around the assumed natural origin of SARS-CoV persists, as no close relatives to SARS-CoV-1 or SARS-CoV-2 have been identified in diverse local animals, including palm civets, from relevant Chinese regions13,14.
If SARS coronavirus had indeed an artificial yeast origin, an important point would be the identification of the putative input progenitor SARS-CoV like nucleotide sequence that went into yeast for assembly. For example, it could be a highly pathogenic virus designed for, or adapted to human cells and subsequently selected for yeast artificial assembly and passage together with some genetic modifications44 of the virus to attenuate its virulence. Indeed, yeast reverse genetics in the context of stable, genetically easily modifiable and scalable virus vaccine production have been described20,45. Then its release back into the human host would likely initiate a rapid succession of complex reversal mutations toward its more pathogenic original structure41,44. Intriguingly, during the first months of the SARS-CoV-2 outbreak, the genomic regions of nsp3 and spike protein had the highest mutational rate within the SARS-CoV-2 genome46 which may have interfered with the yeast homology regions detected in the present study. During an epidemic, such reversal mutations toward an unidentified artificial genotype would be highly detrimental to most public health countermeasures, including pharmacological interventions and vaccinations. In contrast, through specific guidance of countermeasures such as vaccine development, detailed knowledge about the input progenitor’s nucleotide sequence would effectively confer population immunity against the pathogen.
With regard to the most characteristic sequence signature of SARS-CoV-2, Andersen et al.1 questioned the possibility that the polybasic cleavage site at the critical S domain junction was acquired during passage in cell culture. However, according to our data, this cleavage site is specifically compatible with a recombination event including chromosome XIII of S. cerevisiae, which shares a unique nucleotide sequence that encodes the necessary insert PRRA. From a host viewpoint, our results suggest that an artificial origin of both SARS-CoV-2 and SARS-CoV-1 should coincide with an emergence of synthetic yeast lineages unnaturally enriched in their chromosomes, due to recombination, with sequences from these coronaviruses. Arguably, such claim would be testable with sequencing data from laboratory and field samples. Collectively, our results offer a new lead for the further understanding of SARS coronavirus origins.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)