Keywords
lung microbiome, lung virome, metagenomic shotgun sequencing, metagenomic next-generation sequencing, clinical metagenomics, ventilator-associated pneumonia, mechanical ventilation
This article is included in the Genomics and Genetics gateway.
This article is included in the Cell & Molecular Biology gateway.
This article is included in the Pathogens gateway.
lung microbiome, lung virome, metagenomic shotgun sequencing, metagenomic next-generation sequencing, clinical metagenomics, ventilator-associated pneumonia, mechanical ventilation
BAL bronchoalveolar lavage
CMV: cytomegalovirus
COPD: chronic obstructive pulmonary disease
ETA endotracheal aspirate
HSV: Herpes simplex virus
ICU: intensive care unit
mNGS metagenomic shotgun sequencing
PCR: polymerase chain reaction
VAP: ventilator associated pneumonia
The respiratory microbiota is composed of bacteria, viruses, fungi and archaea that interact and constitute a barrier protecting the lungs from colonization by pathogenic microorganisms.1 A dysbiosis of the respiratory microbiota (change in composition or functioning) may be associated with a pathological state and promote infection, as suggested in asthma, chronic obstructive pulmonary disease (COPD) or cystic fibrosis.2 In intensive care units (ICU), some studies have also described an association between a decrease in lung microbiota diversity and the occurrence of ventilator-associated pneumonia (VAP).3 The lung microbiota is indeed deeply affected by different therapies in critically ill patients such as antibiotics or mechanical ventilation.4 A better understanding of its evolution could thus help manage VAP.5 To date, outside lung transplant recipients, studies in ventilated patients have mainly focused on bacterial microbiota. Very few have studied the viral populations besides Papazian et al 10 years ago.6 However, analysis of bacterial fungal and viral communities simultaneously seems essential to obtain a comprehensive view of the potential dysbiosis.7 The human lung virome integrates all viruses present in the lungs, both eukaryotic and prokaryotic viruses (also called phages), and plays a fundamental role in the development and regulation of the innate and adaptive immune system.7,8 For instance, a respiratory syncytial virus (RSV) infection leads to a deregulation of the immune system that persists for several weeks.9 Identification of influenza virus or rhinovirus in the airways has also been associated with the occurrence of bacterial infections.10 Specific analysis of the lung virome in ICU is thus essential to improve our understanding of the VAP pathophysiology.
In a pilot study based on a prospective observational cohort study (January 2015 to June 2016, Paris, France, in press) (ethical authorization CE SRLF 15-41), we aimed to evaluate the feasibility of studying the lung virome in patients under mechanical ventilation in ICU by metagenomic shotgun sequencing (mNGS) using an extraction method of nucleic acids allowing an analysis of the bacterial microbiota on the same samples.
All samples were provided from patients initially included in a cohort study titled “Lung microbiota of ventilated ICU patients: towards a new understanding of nosocomial pneumonia” (in press). The ethics committee of the French intensive care society approved the cohort study, authorizing the use of the samples for the study of bacterial microbiota and of the virome each as a part of this cohort study (reference CE SRLF 15-41, date of acceptance October 1st 2015).
Informed consent was sought on admission to the intensive care unit in mechanically ventilated patients placed under general anaesthesia or in patients who presented signs of acute respiratory failure and required orotracheal intubation. As a result, oral consent was systematically obtained from patient’s next of kin. After patient’s extubation, verbal informed consent was systematically confirmed by the patients themselves except in cases of altered mental status or delirium. According to French regulations on non-interventional studies, the ethics committee of the French Intensive Care Society considered oral consent to be sufficient.
For each patient, when informed consent was confirmed by the patient themselves after extubation, verbal consent for publication was also obtained.
Inclusion criteria in the cohort study were age over 18 years, intubation at admission or within the six hours before, expected duration of mechanical ventilation above 72 hours. Exclusion criteria were a known chronic lung disease (severe chronic obstructive pulmonary disease, emphysema, bronchiectasis, cystic fibrosis, lung transplant or a restrictive lung disease), an invasive ventilation for more than 72 hours in the last six months, an immunosuppression (HIV with less than 200 CD4 lymphocytes/mm3, neutropenia below 500/mm3, treatment by immunosuppressive agents), and absence of consent to participate. Overall, 86 patients were included in the cohort study.
For this feasibility study, we selected nine patients, in order to represent all subgroups of patients (patients who were hospitalized for a bacterial or viral community-acquired pneumonia, and/or patients who developed VAP). The respiratory virome of 24 samples (endotracheal aspirate (ETA) or bronchoalveolar lavage (BAL)) in these 9 representative patients was evaluated.
Samples were removed from -80°C storage and were put in 2 ml lysis matrix tube Y (MP Biomedicals®, catalogue number SGD135.55) for bead beating. For ETA only, 500 microliters of PBS EDTA free with Ca2+ and Mg2+ (Invitrogen®) were added to the lysis matrix tube. Samples were homogenized for 30 seconds two times at 6000 rpm using PlexIDbb instrument (Precellys Bertin technologies®). After centrifugation (one min at 8000 rpm), viral nucleic acid of samples was extracted from each sample using the nucleic extraction platform based on magnetic silica technology NucliSENS® easyMAG® (Biomerieux®). For each nucleic acid extraction, negative controls (NC) (600 μL of PBS EDTA free) were used. For each sample and NC, after Qubit fluorometer quantification® of the nucleic acid amount (Qubit dsDNA Hs Assay Invitrogen®, catalogue number Q32851), DNA and RNA libraries were generated in order to analyze the entire viral population.
DNA libraries were generated using the Nextera DNA XT Kit (Illumina®, San Diego, CA, USA, catalogue number FC-131-1096) in a five steps procedure: i) a methylated DNA removal step using MBD2-Fc beads in order to eliminate human DNA ii) a DNA concentration by Zymo DNA clean concentrator iii) a 5 minute ATM enzyme tagmentation step performed at 55°C with a final enzyme inhibition iv) a final 16 cycles PCR reaction with Illumina® sequencing primer (Nextera XT Index Kit v2 Set D, 96 indexes, catalogue number FC-131-2004).
RNA libraries were performed using RNA Seq Trio kit (Nugen®, San Carlos, California, USA, catalogue number M01440) as per manufacturer’s instructions. This kit is notably used for small amounts of RNA (between 500 picogram and 50 nanogram).11 After DNase incubation (37°C 10 min, 60°C 5 min, hold at 4°C) double strand cDNA was synthetized performing two successive PCR reaction containing 25 μL of eluate after nucleic acid extraction, and PCR products were purified using AMpure magnetic bead-based purification system (Beckman Coulter, Inc, Atlanta, Georgia) as per manufacturer’s instructions.
After single primer isothermal amplification (SPIA), indexed Illumina libraries were prepared as per manufacturer’s instructions. First, cDNA strands fragmentation was performed to obtain homogenous 300 pb PCR products (25°C-30 min, 70°C-10 min, hold at 4°C). Then library amplification of the 300 pb based long cDNA with Illumina® sequencing primer, consisted in two PCR reactions separated by a targeted depletion with Anydeplete® (60°C-30 min, 95°C-5 min, hold at 4°C).
After each library preparation, PCR products were purified using a 0.9 volume of AMpure® magnetic bead-based purification system (Beckman Coulter, Inc, Atlanta, Georgia, catalogue number A63881) as per manufacturer’s instructions and eluted in low Tris-EDTA buffer. Successful amplifications were verified in 4200 Tape Station Agilent bioanalyzer (Agilent Technologies®) as per manufacturer’s instructions, with DNA 5000 screen tape (catalogue number 5067-5588) and reagents (catalogue number 5067-5589) and indexed-DNA sequences quantified in purified samples using a quantitative PCR performed with KAPA Library quantification kit for Illumina platforms (Kappabiosystems®, catalogue number KK4854 – 07960298001) as per manufacturer’s instructions. After sample preparation following the Illumina’s protocol instructions, 24 samples for final DNA library and 24 samples for final RNA library were loaded at 1.8 pM and sequenced in two sequencing runs on NextSeq 500 platform (Illumina®, San Diego, California, USA), using the indexing strategy and 150-bp paired-end reads (V2 300 cycle kit, Illumina® catalogue number MS-102-2002) as per manufacturer’s instructions.
The bioinformatics analysis was performed using the SURPI (Sequence based ultra rapid pathogen identification) computational pipeline12 available at https://chiulab.ucsf.edu/?s=surpi. All the sequences were deposited in the European nucleotide archive: ENA: Analysis of the lung virome under mechanical ventilation by high-throughput sequencing accession number PRJEB56382 (second accession number ERP141316).18
Viruses identified in negative controls were then excluded from the analysis of other samples.
In order to evaluate accuracy and sensitivity of metagenomic shotgun sequencing method, we used conventional targeted polymerase chain reaction (PCR) to identify respiratory viruses in each sample. A respiratory panel of 17 pathogenic viruses (including rhinovirus, influenza and parainfluenza virus and syncytial respiratory virus) was investigated using FilmArray® Respiratory Panel 2 (bioMérieux, France) as per manufacturer’s instructions, on the first sample collected for each patient.13 Herpes simplex viruses and cytomegalovirus were also tested by quantitative PCR (RealStar® HSV PCR Kit CE - Altona-Diagnostics catalogue number 061013, CMV RealTime Abbott, Abbott Molecular® catalogue number 05N23-090) in BAL samples as per manufacturer’s instructions.
The analysis code used in this study has been deposited in Zenodo.20
In total, nine patients were included in this study, all males with a median age of 72 years [66;72], 33% active smokers, with a median duration of mechanical ventilation of 18 days [16;20]. The patient’s characteristics are presented in Table 1.19
Overall, eight of these patients presented a community acquired pneumonia (CAP) or an aspiration pneumonia on ICU admission, two presented a viral pneumonia and three of them developed a VAP during their ICU stay (patients 1, 2 and 3). Data from conventional microbiology for all subtypes of bacterial pneumonia are presented in Table 2 with five out of eight having documented CAP (Table 2).
Four viral species were identified by conventional PCR methods among six patients: patient 1 with influenza virus B (Orthomyxoviridae), patients 2 and 3 with cytomegalovirus (Herpesviridae), patients 4 and 5 with respiratory syncytial virus (Paramyxoviridae), and patient 9 with influenza virus A (Orthomyxoviridae). No viral species were identified by PCR in patients 6, 7 and 8.
Metagenomic shotgun sequencing generated a total of about 1 billion reads, a majority of them corresponding to human reads (83%) for RNA and DNA libraries (Figure 1a and b). Nevertheless, SURPI identified a total of 196,580 viral reads. After family assignation and consideration of positive detection in samples with at least five reads of the same species, eleven viral families were detected, including seven families of eukaryotic viruses and four families of phages (Figure 2).
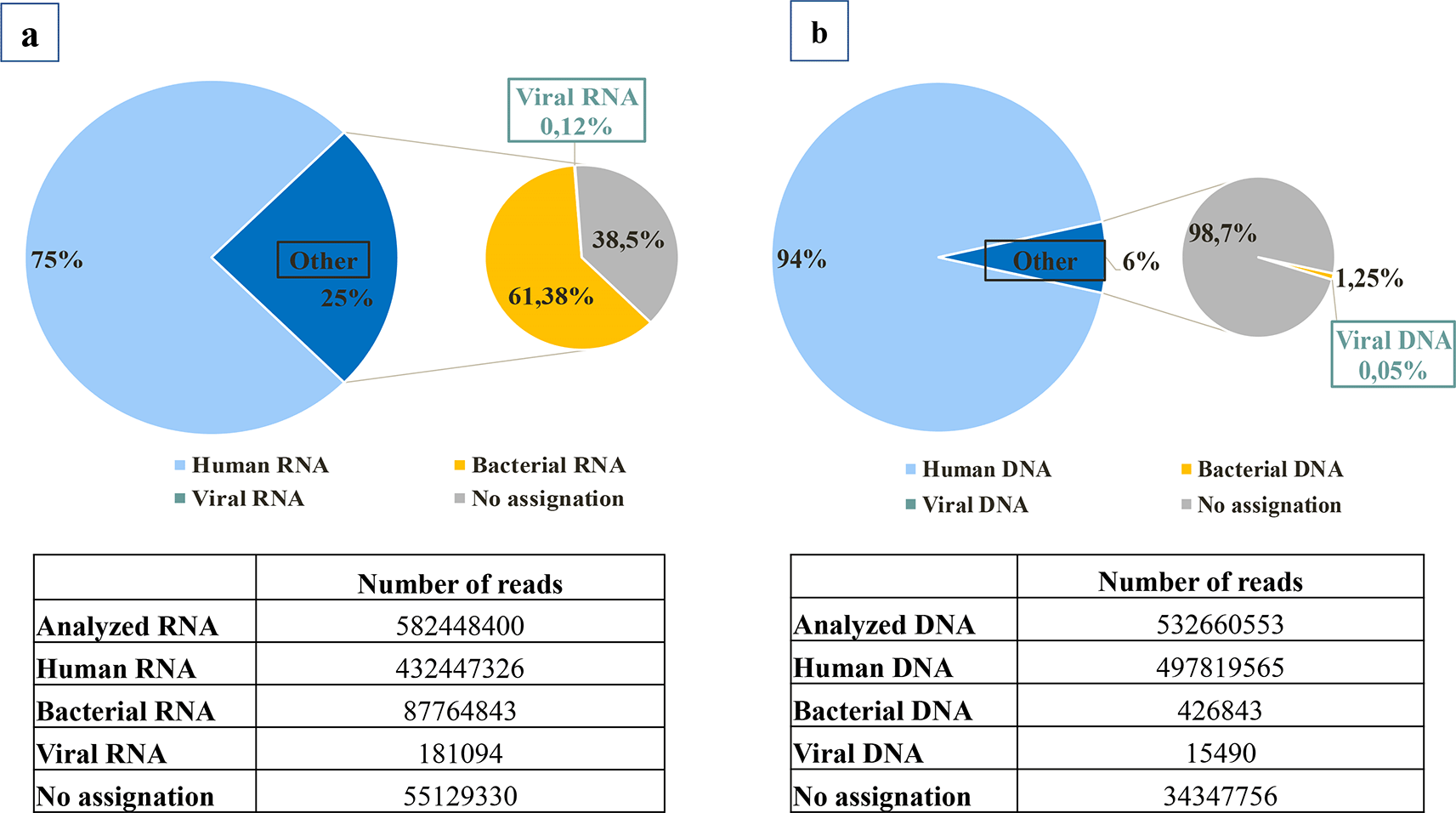
Assignation of reads obtained after high throughput sequencing are represented in (a) for RNA libraries and (b) for DNA libraries. For RNA library, 75% were human RNA sequences and 25% were classified as “other”. For DNA library, 94% were human DNA sequences and 6% were classified as “other”. For both types of libraries, the remaining sequences corresponded to bacterial, viral, or unassigned sequences expressed as a percentage of non human sequences. Viral sequences represented only 0.12% of the other non-human RNA sequences and 0.05% of the other non-human DNA sequences.
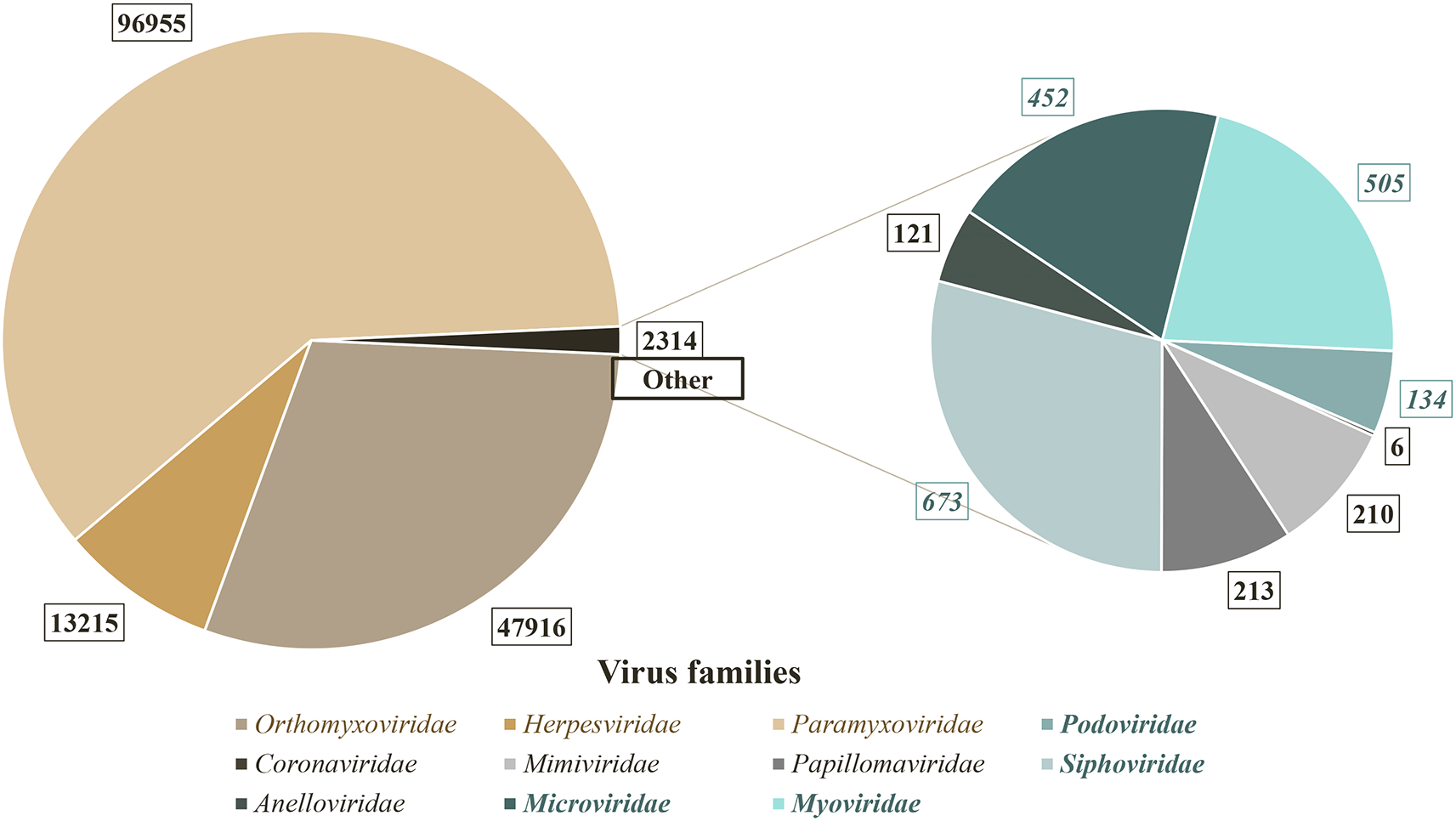
Overall, 11 virus families have been identified after high throughput sequencing. The number of reads assigned to each family are shown on the diagrams. The three main families are Orthomyxoviridae, Herpesviridae and Paramyxoviridae. Only 2314 reads belonged to other families. Families of eukaryotic viruses are represented in grey or brown tones. Families of phages are represented in green tones and in bold.
Virus detection per patient is given in Table 3. All viruses detected by conventional targeted PCR were identified by mNGS. In addition, a large number of viruses not detected by PCR were detected by metagenomic shotgun sequencing. For patients 1 and 9, the multiplex PCR identified influenza virus and mNGS identified nine different viral families including influenza virus, Herpesviridae and Anelloviridae, and phages. The same observation was made for patients 2 and 3 in whom cytomegalovirus (CMV) was detected by specific PCR and by mNGS in addition to many families of bacteriophages. For patients 4 and 5, while respiratory syncytial virus was detected by both PCR and mNGS, the latter method also identified a significant number of other viruses such as Herpesviridae (mostly represented by Herpes simplex virus 1 (HSV1)) and Anelloviridae. For patients 6, 7 and 8, while no virus was identified by PCR, numerous viral families were identified by mNGS.
The presented method enabled to determine the respiratory virome profile with a good accuracy compared to multiplex PCR. Interestingly we were able to describe the bacterial microbiota on the same samples, as shown in our previous pilot study on respiratory bacterial microbiota, using the same nucleic acid extraction before 16s rRNA high-throughput sequencing.14 Overall, these results demonstrated the opportunity to study both bacterial and viral microbiota in respiratory samples of ventilated patients, providing a more comprehensive view of the respiratory microbiome and thus allowing a better understanding of VAP pathophysiology.15 The small number of samples and patients do not allow generalization at this stage. As previously observed in other studies, the number of viral reads is lower than those related to human or bacterial genomes by suing global metagenomics without a dedicated approach to enrich in viral particles.16,17 However this did not hamper the overall sensitivity as all respiratory viruses identified by targeted multiplex PCR were also detected by metagenomics.
This feasibility study thus validates the strategy of a unique metagenomics protocol with the final aim of studying not only the lung bacterial microbiota but also the lung virome.15 Adding the ITS2 high-throughput sequencing, it will allow investigating global respiratory microbiota profiles and coevolution including bacteria, fungi and viruses in ventilated patients. Larger cohort studies are needed to fully characterize pulmonary dysbiosis in ventilated patients who have developed a VAP to understand whether pulmonary dysbiosis is a cause, a consequence or both.
VAP prevention is a crucial challenge for the management of ICU patients. Obviously in case of causal pulmonary dysbiosis, a better understanding of infectious steps leading to VAP development can help to define targeted interventions on the bacterial microbiota, the mycobiota and the virome.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)