Keywords
hagfish, cyclostome, whole genome assembly, gene prediction
This article is included in the Genomics and Genetics gateway.
This article is included in the Japan Institutional Gateway gateway.
hagfish, cyclostome, whole genome assembly, gene prediction
Extant jawless fishes (cyclostomes) are divided into two groups, hagfishes (Myxiniformes) and lampreys (Petromyzontiformes).1 They have been studied from various viewpoints mainly because they occupy an irreplaceable phylogenetic position among the extant vertebrates, having diverged from all other vertebrates during the early Cambrian period. Even after massive efforts of whole genome sequencing for invertebrate deuterostomes,2,3 genome-wide sequence information for species in this irreplaceable taxon was limited until the genome analyses for two lamprey species, the sea lamprey Petromyzon marinus and the Arctic lamprey Lethenteron camtschaticum, which were published in 2013.4,5
In parallel, biological studies involving individual genes have been conducted for both lampreys and hagfishes. Developmental biologists, in particular, have largely relied on lampreys whose embryonic materials are accessible through artificial fertilization,6 whereas studies on hagfishes have been limited to non-embryonic materials, with a few notable exceptions.7–9 This type of molecular biological study is expected to be more thoroughly performed if a comprehensive catalogue of genes is available. For lampreys, derivation of a reliable comprehensive gene catalogue was long hindered by the peculiar nature of protein-coding sequences, which are characterized by high GC-content, codon usage bias, and biased amino acid compositions.5,10,11 To reinforce existing resources for lampreys, we previously performed a dedicated gene prediction for L. camtschaticum12 and provided a gene catalogue with comparable or superior completeness to other equivalent resources.4,13
As of July 2022, no whole genome sequence information is available for hagfishes except for the one at Ensembl13 that remains unpublished, a fact that hinders the comprehensive characterization of gene repertoires and their expression patterns. Currently, some efforts for genome sequencing and analysis are ongoing that aim to resolve large-scale evolutionary and epigenomic signatures,9 inspired partly by the relevance of hagfish to understanding patterns of whole genome duplications14–19 and chromosome elimination.20–22 In contrast to those efforts, which are necessarily targeting reconstruction of the genome at chromosome scale, in this study we aimed at providing a data set covering as many full-length protein-coding genes as possible, to enable gene-level analysis on molecular function and evolution of hagfishes, an indispensable component of the vertebrate diversity.
A 48cm-long adult male individual of Eptatretus burgeri caught at the Misaki Marine Station in June 2013 was used for the study. After anesthetization in 1% tricaine and decapitation, the testis was sampled from which genomic DNA was extracted with the conventional phenol/chloroform extraction method,23 and the genome sequencing was performed as outlined in Figure 1. The study was conducted with all efforts to ameliorate any suffering of animals, in accordance with the institutional guideline Regulations for the Animal Experiments by the Institutional Animal Care and Use Committee (IACUC) of the RIKEN Kobe Branch. The extracted genomic DNA was sheared using an S220 Focused-ultrasonicator (Covaris), which allowed us to retrieve DNA fragments of variable length distributions. Table 1 includes detailed information on amounts of starting DNA as well as conditions for shearing. The sheared DNA was subjected to paired-end library preparation using the KAPA LTP Library Preparation Kit (KAPA Biosystems). The optimal number of PCR cycles for library amplification was determined by quantitative PCR based on SYBR Green, using the KAPA Real-Time Library Amplification Kit (KAPA Biosystems) with Illumina library compatible primers (5′-AATGATACGGCGACCACCGA-3′ and 5′-CAAGCAGAAGACGGCATACGA-3′), and an aliquot of adaptor-ligated DNA,24 at 98°C for 45 sec followed by 25 cycles of amplification at 98°C for 15 seconds, 60°C for 30 seconds, and 72°C for 30 seconds, in ABI 7900HT Real Time PCR system (Thermo Fisher Scientific). The optimal Ct value was determined in SDS 2.4 software (Thermo Fisher Scientific) as cycles that reaches FS1 (Fluorescent Standard 1) but does not exceed FS2 (Fluorescent Standard 2). Libraries were further size selected using Agencourt AMPure XP (Beckman Coulter). Mate-pair libraries were prepared using the Nextera Mate Pair Sample Prep Kit (Illumina), employing our customized iMate protocol.25 Detailed information of the paired-end and mate-pair libraries are described in Table 1. Libraries were quantified using the KAPA Library Quantification Kit (KAPA Biosystems) and sequenced on HiSeq 1500 (Illumina) operated by HiSeq Control Software v2.0.12.0 using HiSeq SR Rapid Cluster Kit v2 (Illumina) and HiSeq Rapid SBS Kit v2 (Illumina), or on HiSeq X (Illumina) operated by HiSeq Control Software v3.3.76, or on MiSeq operated by MiSeq Control Software v2.3.0.3 using the MiSeq Reagent Kit v3 (600 Cycles) (Illumina). Read lengths were 127 or 251 nt on HiSeq 1500, 151 nt on HiSeq X, and 251 nt on MiSeq. Base calling and generation of fastq files were performed with RTA v1.17.21.3 (Illumina, RRID:SCR_014332) and bcl2fastq v1.8.4 (Illumina, RRID:SCR_015058) for the sequencing data of HiSeq 1500 and MiSeq, or by RTA v2.7.6 and bcl2fastq v2.15.0 for the sequencing data of HiSeq X. Illumina adaptor sequences and low-quality bases were removed from the paired-end sequencing reads by Trim Galore v0.3.3 (RRID:SCR_011847) with the ‘--stringency 2 --quality 30 --length 25 --paired --retain_unpaired’ options. Mate-pair reads were processed to identify the junction adaptor by NextClip v1.126 (RRID:SCR_005465) with the default parameters.
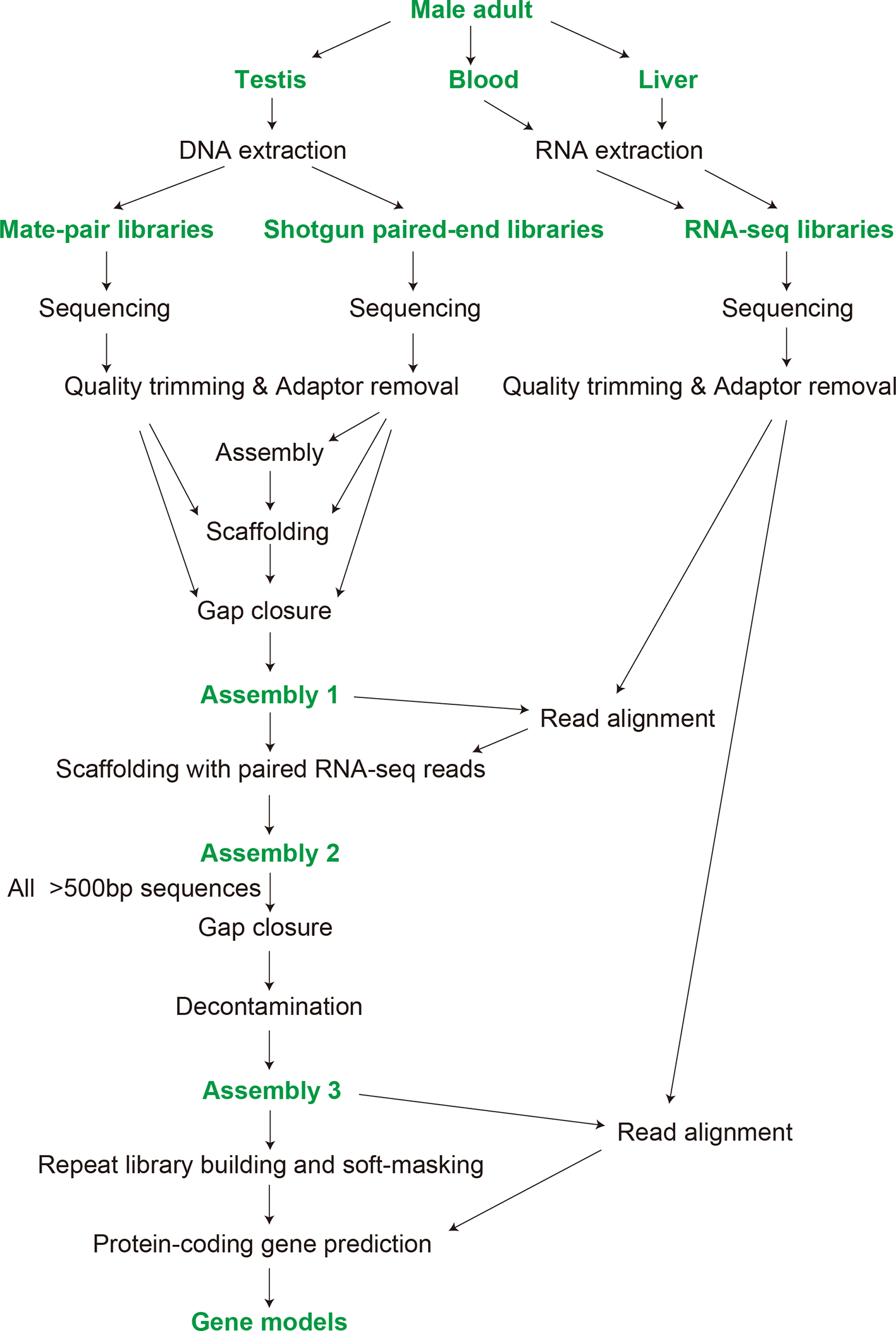
Samples, raw data, and products are indicated with green letters, while computational steps are labelled in black. See Methods for the details including the choice of the programs used in individual computational steps.
| C. RNA-seq libraries | |||||
|---|---|---|---|---|---|
| Accession ID | Library ID | Tissue | Amount of total RNA used (μg) | # PCR cycles | # read pairs |
| DRX218831 | P238_01_1 | Liver | 1 | 6 | 55,675,220 |
| DRX218832 | P238_02_1 | Blood | 1 | 5 | 57,600,815 |
Total RNAs were extracted from the liver tissue and the blood of the above-mentioned adult individual with Trizol reagent (Thermo Fisher Scientific) following the manufacturer’s instruction. The RNA was treated with DNase I to digest genomic DNA. Quality control was performed with Bioanalyzer 2100 (Agilent Technologies), which yielded the RIN values of 8.7 and 9.1 for the respective tissues. Libraries were prepared with TruSeq Stranded mRNA LT Sample Prep Kit (Illumina).27 The amount of total RNA used for library preparation and the number of PCR cycles applied for library amplification are described in Table 1 and Figure 2. Removal of Illumina adaptor sequences and low-quality bases was performed with Trim Galore v0.3.3 as outlined above. Alignment of the RNA-seq reads to the genome assembly was performed by HISAT2 v2.2.128 (RRID:SCR_015530) with the options ‘-k 3 -p 20 --pen-noncansplice 1000000’.
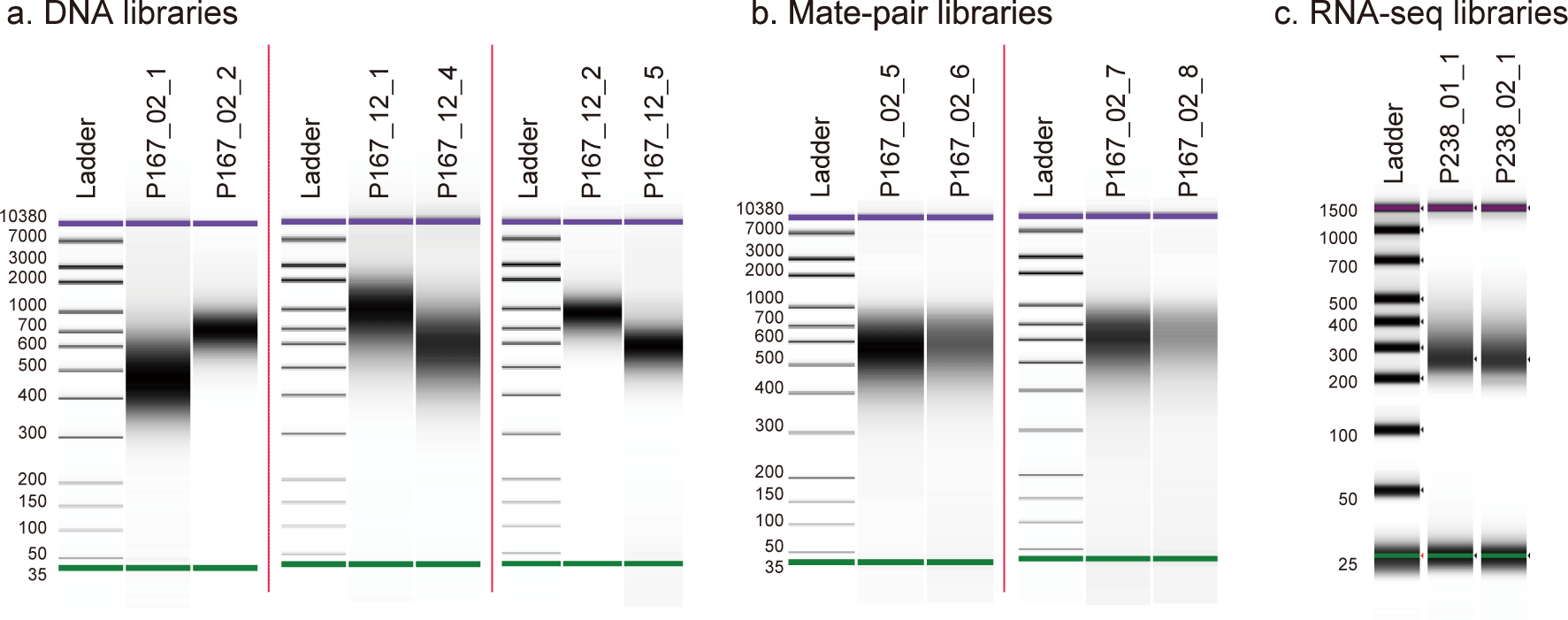
a, Shotgun DNA libraries analyzed by Bioanalyzer High Sensitivity DNA Kit (Agilent). b, Mate-pair libraries analyzed by Bioanalyzer High Sensitivity DNA Kit. c, RNA-seq libraries analyzed by TapeStation High Sensitivity D1000 ScreenTape Assay Kit (Agilent).
De novo genome assembly and scaffolding of Illumina short reads processed as described above were performed by the program PLATANUS v1.2.429 (RRID:SCR_015531) with its default parameters. The assembly employed paired-end reads and single reads whose pairs had been removed for quality filtering, and the scaffolding employed paired-end and mate-pair reads. The gap closure employed all of the single, paired-end, and mate-pair reads after processing. The obtained sequences were further scaffolded with paired-end RNA-seq reads with the program P_RNA_Scaffolder30 (commit 7941e0f on May 30, 2019, at GitHub) with the options ‘-s yes -b yes -p 0.90 -t 20 -e 100000 -n 100’, followed by another gap closure run with PLATANUS ‘gap_closure’ using the same set of reads used in the above-mentioned gap closure run. The resultant genomic sequences were further screened for the species’ own mitochondrial DNA fragments, contaminating organismal sequences, PhiX sequences loaded as a control in the Illumina sequencing system, and sequences shorter than 500 bp, as performed previously.31
To obtain species-specific repeat libraries, RepeatModeler v1.0.832 (RRID:SCR_015027) was executed with its default parameters. Repeat element detection in the genome sequence was performed by RepeatMasker v4.0.533 (RRID:SCR_012954), which employs the National Center for Biotechnology Information (NCBI) RMBlast v2.2.2734 (RRID:SCR_022710), using the custom repeat library obtained above by RepeatModeler. Genomic regions detected as repeats were soft-masked by RepeatMasker with the ‘-nolow -xsmall’ options.
Construction of gene models was performed by employing the gene prediction pipeline BRAKER v2.1.435 (RRID:SCR_018964) with the options ‘--min_contig=500 --prg=gth --softmasking --UTR=off’ (Figure 1). This computation employed RNA-seq read alignments in BAM files onto the genome assembly and a set of peptide sequences prepared as follows. The set of peptide sequences used as homolog hints included the predicted proteins of the Arctic lamprey (34,362 sequences, previously designated as GRAS-LJ12), which were aligned to the soft-masked genome assembly.
Our technical procedure employing the genome assembly program PLATANUS29 that previously produced genome assemblies for multiple shark species with modest investment36 yielded genome sequences consisting of 4,519,897 scaffolds (Assembly 1 in Figure 1) with an N50 length of 238 Kbp (length cutoff=500 bp). To improve the continuity of fragmentary sequences that were derived from transcribed regions but were separated from exons, the sequences in Assembly 1 were further scaffolded with paired-end RNA-seq reads, which resulted in 4,505,643 sequences (Assembly 2) with an N50 length of 264 Kbp (length cutoff=500 bp). These sequences were filtered for the length of >500 bp, processed again for gap closure with the program PLATANUS, and scanned for contaminants of microbes and artificial oligos used for sequencing. Through this procedure, we have obtained 114,941 sequences with the minimum and maximum lengths of 500 bp and 2.064 Mbp, respectively, marking the N50 scaffolding length of 293 Kbp (Assembly 3).
Using the resultant genome sequences (Assembly 3), genome-wide prediction of protein-coding sequences were performed with the program pipeline BRAKER.35 After preliminary runs with variable parameters and input data sets, we conducted a prediction run with transcript evidence and peptide hints, which resulted in a set of 46,295 genes, with the maximum length of the putative peptides of 19,580 amino acids. These sequences have systematic identifiers Eptbu0000001–Eptbu0046295 with suffixes ‘.t1’–‘.t6’ depending on the multiplicity of predicted peptide variants derived from alternative splicing. These sequences are available under https://figshare.com/projects/eburgeri-genome/77052.37–41
To confirm the coverage of the genome assembly, paired-end RNA-seq reads were aligned to the genome sequence (Assembly 3) with splicing-aware read mapping program HISAT2 as described in the Methods section. This computation resulted in mapping of the reads to the nuclear and the mitochondrial genome sequences of E. burgeri at high proportion, at 91.64% and 5.17% respectively.
It has been previously shown that completeness scores of cyclostome genomes tend to be underestimated, when their rapid-evolving nature and phylogenetic position is not taken into consideration.27 In this study, completeness of the genome assemblies was assessed with CEGMA v2.542 (RRID:SCR_015055) and BUSCO v2.0.143 (RRID:SCR_015008). For both CEGMA and BUSCO, we employed not only the reference gene sets provided with these pipelines but also the core vertebrate genes (CVG) that was developed specifically for vertebrates from isolated lineages such as elasmobranchs and cyclostomes.27 The completeness assessments executed using CEGMA and CVG on the gVolante webserver44,45 returned percentages of single-copy orthologs detected as ‘complete’ of 65%, and ‘complete or partial/fragmented’ of 91%. Use of BUSCO v2.0.143 with CVG resulted in the detection of 'complete' single-copy orthologs of 83.7%, and ‘complete or partial/fragmented’ single-copy orthologs of 93.6% (Table 2). The difference of the completeness scores between the assessments of the genome assembly and the gene models might be explained by decreased sensitivity of detecting divergent multi-exon genes in the genome. Altogether, the resultant set of gene models is expected to encompass more than 90% of the protein-coding genes in the E. burgeri genome.
| Species | Source | # Genes (# Peptides) | Maximum peptide length (amino acids) | Completeness scoreb (%) | |
|---|---|---|---|---|---|
| Only ‘Complete’ | Including ‘Fragmented’ | ||||
| Eptatretus burgeri | This study | 46295 (50127) | 19580 | 83.7 | 93.6 |
| Lentheteron camtschaticum | GRAS-LJ12,a | 34435 | 19612 | 90.1 | 98.7 |
| Petromyzon marinus | PMZ_v3.019 | 20940 (20950) | 18818 | 57.1 | 89.3 |
| Petromyzon marinus | Ensembl gene build13 | 10415 (11442) | 18900 | 84.1 | 94.9 |
| Petromyzon marinus | PMZ1.05 | 24132 (24271) | 17467 | 63.5 | 89.3 |
This data set is oriented towards gene-level analysis including phylogenomic analysis and phylome exploration aiming at studying gene family evolution, rather than the analysis of complete genome structure. Importantly, the total length of the genome sequences obtained in this study amounts only to approximately 1.7 Gbp which is smaller by more than 1 Gbp than the genome size estimate based on flow cytometry of nuclear DNA content21 (2.91 Gbp). For investigating the structural evolution of the whole genome, such as chromosome elimination or large-scale synteny conservation, it may be advisable to wait for other resources to be released without embargo.
The obtained gene models sometimes include multiple transcripts and their deduced amino acid sequences per gene, because of predicted alternative splice variants. For use in phylogenomics and ortholog clustering, a set of amino acid sequences without splice variants (doi: 10.6084/m9.figshare.11971932)37 has also been made available. These sequence data are available for BLAST searches on the Squalomix project site (https://transcriptome.riken.jp/squalomix/).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)