Keywords
Monoterpene indole alkaloids, Catharanthus roseus, Apocynaceae
This article is included in the Nanopore Analysis gateway.
This article is included in the Plant Science gateway.
This article is included in the Genomics and Genetics gateway.
Monoterpene indole alkaloids, Catharanthus roseus, Apocynaceae
The Madagascar periwinkle, Catharanthus roseus (L.) G. Don, is an Apocynaceae plant native to Madagascar. C. roseus produces several specialized metabolites including monoterpene indole alkaloids (MIA; O’Connor and Maresh, 2006). These molecules are produced by plants to face biotic and abiotic pressures accounting for their wide range of bioactive properties (Dugé de Bernonville et al., 2015). Above all, MIAs produced by C. roseus are well-known for being part of the human pharmacopoeia against cancer, such as the well-known vinblastine and vincristine, and other MIA derivatives, including vinorelbine (O’Connor and Maresh, 2006).
Due to its high economic importance, C. roseus has extensively been studied within the last three decades becoming the model species for MIA biosynthetic pathway studies (see Pan et al., 2016 and Kulagina et al., 2022 for extensive review). C. roseus genome was firstly sequenced in 2015 (Kellner et al., 2015). Recently, a more contiguous version (v2) was generated to ease inter-species genomic comparison (Franke et al., 2019). To date, C. roseus genome sequencing and assembly did not benefit from the development of third generation sequencing technologies that lead to more contiguous genome (Jiao and Schneeberger, 2017). Thanks to these new technologies, we present here an even more contiguous genome assembly. This updated version (v2.1) should ease inter-species studies in order to better understand the diversification of MIAs and the evolution of their biosynthetic pathways.
C. roseus cv ‘SunStorm® Apricot’ seeds (variety ID: 70001114, Syngenta flowers, Basel, Switzerland) were greenhouse-grown at the University of Tours for 1 month before sampling. DNA was extracted from C. roseus leaves using Qiagen Plant DNeasy kit (ID: 69204, Qiagen, Hilden, Germany) following the manufacturer’s instructions. Illumina sequencing library were constructed using the TruSeq DNA PCR-free kit (ID: 20015962, Illumina, San Diego, USA) and sequenced in paired-end mode (2 × 150 bp) by Eurofins Genomics (Les Ulis, France) using Illumina NextSeq500 technology. Future Genomics Technologies (Leiden, The Netherland) constructed ONT library using ONT 1D ligation sequencing kit (SQK-LSK109, Oxford Nanopore Technologies Ltd, Oxford, United-Kingdom) subsequently sequenced on Nanopore GridION flowcell and Nanopore PromethION flowcell (Oxford Nanopore Technologies Ltd, Oxford, United-Kingdom) with the GuPPy (RRID:SCR_022353) version 3.2.6 high-accuracy basecaller. A total of 114,329,683 paired-end reads were obtained from the Illumina HiSeq sequencing, 908,999 and 2,588,997 from the ONT GridION and ONT PromethION sequencing, respectively.
The C. roseus genome was assembled by Future Genomics Technologies (Leiden, The Netherlands). After adapters removal using Porechop (RRID:SCR_016967) (Wick et al., 2017), ONT reads were first assembled into contig using Flye (RRID:SCR_017016) assembler (v.2.5, Kolmogorov et al., 2019) with the following options: --min-overlap 10000 -i 2. Redundant contigs were removed using Purge_haplotigs (RRID:SCR_017616) (v.1.1.0) followed by two rounds of polishing with Illumina paired-end reads using Pilon (RRID:SCR_014731) (v.1.23, Walker et al., 2014).
RNA-seq data were retrieved from the NCBI Sequence Read Archive (SRA) (RRID:SCR_004891) database using the following accession numbers: ERS1229288, ERS1229289, ERS1229290, ERS1229291, ERS1229292, ERS1229293, ERS1229294, ERS1229295, ERS1229296, ERS1907920, ERS2396963, ERS2396964, ERS2396965, ERS2396966, SRR20661631. These data were individually aligned to the C. roseus genome using HISAT2 (RRID:SCR_015530) (v.2.2.1, Kim et al., 2019). Transcripts were subsequently assembled using the resulting RNA-seq alignments and StringTie (RRID:SCR_016323) (v.2.1.7, Pertea et al., 2015). These individual transcriptomes were further merged using stringtie-merge to a non-redundant set of transcripts. A combination of similarity search using BLASTX (RRID:SCR_001653) and BLASTP (v.2.6.0-1, Camacho et al. 2009) against UniProt (RRID:SCR_002380) database (v.2022-10-12) and hmmscan (v.3.1b2, Finn et al., 2011) against the Pfam (RRID:SCR_004726) database was used to assign putative function to each gene model.
The stat program from BBmap (RRID:SCR_016965) tool (v.38.94, Bushnell, 2014) was used to assess assembly quality. Benchmarking Universal Single-Copy Orthologs (BUSCO v.5.2.2, Simão et al., 2015) (RRID:SCR_015008) with default settings was used to assess genome and gene models completeness using a plant-specific database of 2,326 single copy orthologs (eudicots_odb10). The agat_sp_statistics perl script from the AGAT package (v.0.8.0, Dainat et al., 2022) was used to get the gene models statistics.
Identification and annotation of transposable elements was determined using extensive de novo TE annotator (EDTA v.1.9.5, Ou et al., 2019) (RRID:SCR_022063) using the sensitive mode. This pipeline annotates long-terminal repeat (LTR) using LTR_Finder (RRID:SCR_015247) (v. 1.07, Xu and Wang, 2007) and LTRharvest (RRID:SCR_018970) included in GenomeTools (RRID:SCR_016120) (v.1.5.10, Ellinghaus et al., 2008); terminal inverted repeat (TIR) using Generic repeat finder (v.1.0, Shi and Liang, 2019) and TIR-learner (v.2.5, Su et al., 2019); and Helitrons using HelitronScanner (v.1.1, Xiong et al., 2014). TE size thresholds are further used to prevent false discoveries. Hence, TIR shorter than 80 bp as well as LTR and Helitrons shorter than 100 bp are considered as tandem repeats and short sequences. To prevent false LTR discoveries, LTR are further filtered using LTR_retriever (RRID:SCR_017623) (v.2.9.0, Ou and Jiang, 2018). TIR candidates are classified as MITEs if not exceeding 600 bp. TIR and Helitrons are further filtered using EDTA advanced filters (see Ou et al., 2019 for details). The genome is then masked using the obtained TE library. Unmasked part of the genome is then scanned by RepeatModeler (RRID:SCR_015027) (v.2.0.1, default parameters, Flynn et al., 2020) to identify non-LTR retrotransposons and unclassified TE missed by structure-based TE identification tools. Finally, EDTA uses the provided CDS sequences to remove gene-related sequences.
C. roseus genome was assembled from ONT long-reads using Flye (v.2.5) resulting in a 651.9 Mb assembly distributed across 788 contigs. This assembly was collapsed using purge_haplotigs into 173 scaffolds reducing length to 585,8 Mb but increasing N50 from 10.3 Mb to 12.3 Mb. Assembly polishing was performed twice using Illumina short-reads with pilon (v. 1.23). C. roseus final assembly consisted in 173 scaffolds with a total length of 581.45 Mb. Even though C. roseus v.2.1 displayed similar BUSCO scores compared to C. roseus v.2 based on Eudicotyledons Benchmarking Universal Single-Copy Orthologs (BUSCO), this new version v.2.1 turns out to be much more contiguous with a 12 time less contigs and a six-fold larger N50 (Table 1) (Cuello et al., 2022).
| Version | Assembly size (Mb) | No. of scaff.a | N50 (Mb) | BUSCO scores (genome mode) C [S; D]; F; Mb | Protein coding genes | Ref. |
|---|---|---|---|---|---|---|
| C. roseus v.2 | 541.13 | 2,090 | 2.58 | 97.0 [95.5; 1.5]; 1.3; 1.7 | 34,363 | Franke et al., 2019 |
| C. roseus v.2.1 | 581.45 | 173 | 12.2 | 97.1 [94.2; 2.9]; 1.0; 1.9 | 21,061 | This study |
RNA-seq based gene model prediction using publicly available data resulted in a total of 21,061 genes. Despite less genes were annotated; a higher BUSCO score was obtained (Figure 1). The combination of BLASTP and BLASTX against UniProt database and hmmscan against the PFAM database led to the functional annotation of 76.5% of the predicted genes (16,118 of the 21,062 genes, Supplementary Table S1 in Underlying data (Cuello et al., 2022)). All functionally validated MIA biosynthetic genes from C. roseus could be found in this new version v.2.1 of the genome with identity and coverage percentage ranging from 95 to 100% and 94 to 100%, respectively, with the exception of G10H and DAT (Supplementary Table S2-S3 in Underlying data (Cuello et al., 2022)).
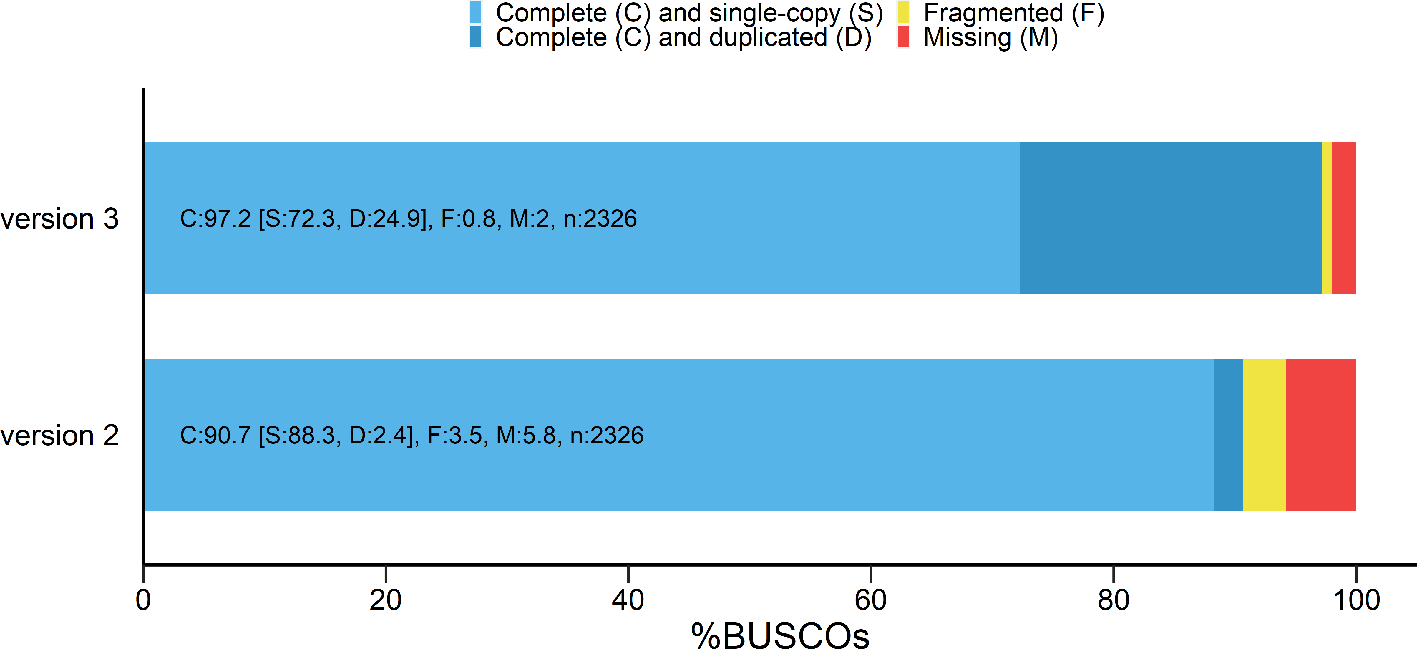
BUSCO: Benchmarking Universal Single-Copy Orthologs.
Finally, we analyzed TE composition of this updated C. roseus genome. While 38.78% of the genome consisted in TE in C. roseus v.2, a higher proportion (42.87%) was annotated as TE in this new version (v.2.1) with similar distribution across the different TE families (Figure 2). It is worth noting that TE proportion of this v.2.1 is closer to the one in its recently sequenced closely related species Vinca minor (Stander et al., 2022).
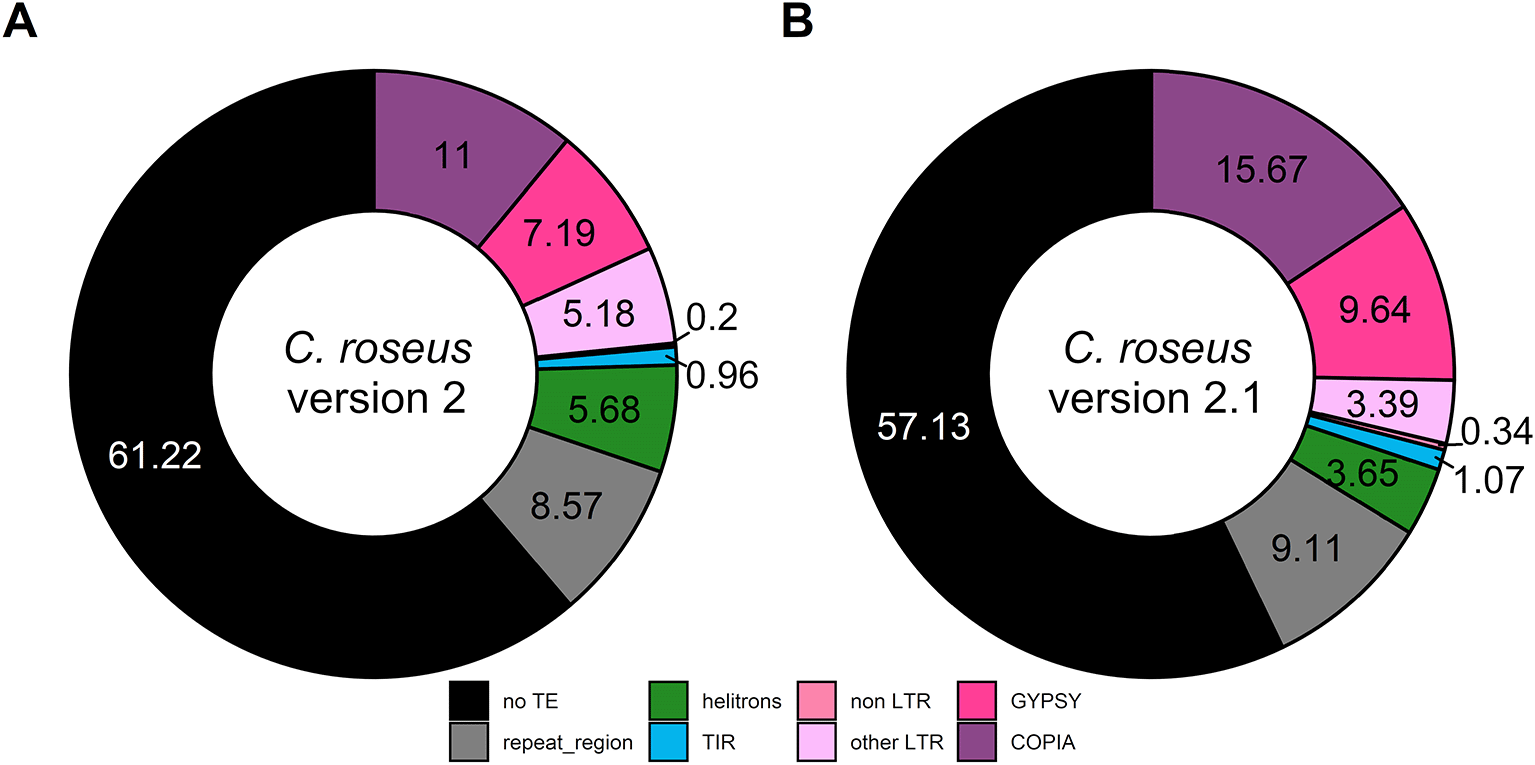
TIR: terminal inverted repeat, LTR: long terminal repeat, non LTR: retrotransposons without LTR sequence, other LTR: LTR containing retrotransposons except for Gypsy and Copia.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)