Keywords
microRNA, transfer RNA-derived fragments, piRNA, circadian, diurnal, small RNA, aging
This article is included in the Cell & Molecular Biology gateway.
This article is included in the Circadian Clocks in Health and Disease collection.
microRNA, transfer RNA-derived fragments, piRNA, circadian, diurnal, small RNA, aging
Diurnal transcript expression is controlled by daily light-dark cycles or by the circadian clock, a conserved molecular feedback loop that regulates daily physiological and behavioral rhythms and is altered during normal aging (Popa-Wagner et al. 2017; Rakshit et al. 2012). Changes in the expression of the diurnal transcriptome with age, including loss and gain of 24-hour rhythmicity, have been shown in multiple organisms, including fruit flies (Kuintzle et al. 2017), zebrafish (Park and Belden 2018), and humans (Chen et al. 2016). While age-related changes in gene expression may impact health and longevity (Acosta-Rodriguez et al. 2021), the causes of shifts in the diurnal transcriptome are not understood. In young flies, transcript rhythmicity is regulated in part by post-transcriptional mechanisms, including regulation by microRNAs (Xue and Zhang 2018). MicroRNAs (miRNAs, miRs) are a class of small (~22 nucleotide) regulatory RNA molecules that cause degradation or translational repression of target mRNAs, altering their level of expression in the cell (Bartel 2018). However, it is not known whether diurnal microRNA profiles change with age and, if so, whether such changes may be involved in altering diurnal transcriptome profiles in old organisms. Here, we address these questions in the fruit fly Drosophila melanogaster.
Metazoan microRNAs are transcribed as primary transcripts (pri-microRNA) that fold into a hairpin structure, are cleaved into a pre-microRNA by the nuclear enzyme Drosha, exported into the cytoplasm, and processed into a microRNA duplex by the enzyme Dicer (Figure 1A) (Bartel 2018). The two strands of the duplex are separated into 5' and 3' arms, one of which is a generally more highly-expressed functional microRNA (miR), and one of which is a largely non-functional product known as the “miR star” (miR*) (Bartel 2018). The functional miR is bound by the protein Ago1 and incorporated into the RNA-induced silencing complex (RISC), while the miR* is degraded in the cell (Bartel 2018). The RISC binds a sequence in the 3' untranslated region (UTR) of the target mRNA with partial base pair complementarity to the seed region (nucleotides 2–8) of the microRNA, causing degradation or translational repression of the mRNA (Bartel 2018).
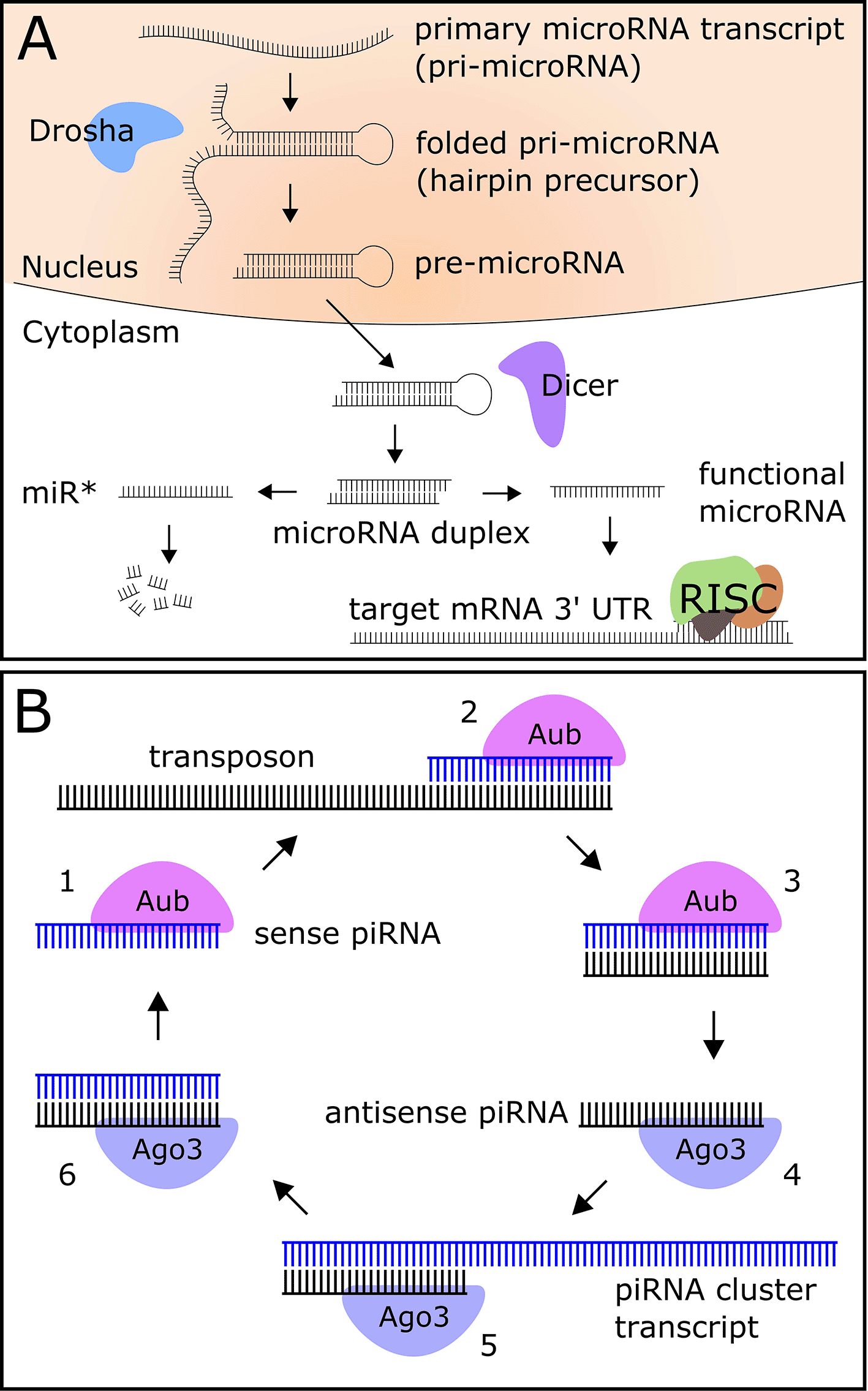
MicroRNAs are well-studied in Drosophila, and several experiments have provided evidence for the role of microRNAs in circadian and light-activated regulation of gene expression in young flies. Yang et al. demonstrated circadian regulation of miR-263a and miR-263b (Yang et al. 2008), while miR-124 has been shown to modulate phase locomotor activity, a circadian clock output behavior (Zhang et al. 2016). Other microRNAs target core circadian clock genes Clk (Kadener et al. 2009) and cwo (Chen et al. 2014) (bantam and let-7, respectively). Further, miR-276a, which targets the core clock gene tim, was shown to be indirectly light-activated (Chen and Rosbash 2016). A miR-959-964 cluster, which shows strong diurnal oscillations, regulates the phase of feeding and immune function (Vodala et al. 2012).
Although microRNAs are known regulators of development (Chandra et al. 2017), they also play important roles in aging and neurodegeneration. For example, miR-34 expression increases in the Drosophila brain during normal aging, and it is involved in the regulation of neurodegeneration and lifespan (Liu et al. 2012). The co-transcribed microRNAs miR-125, let-7 and miR-100 also increase in expression with age, with miR-125 and let-7 required for normal lifespan (Chawla et al. 2016). Many other studies have revealed roles for individual microRNAs in aging and neuroprotection (reviewed in (Kinser and Pincus 2020)).
Another class of Drosophila small RNA that may have a role in aging and neurodegeneration is Piwi-interacting RNAs (piRNAs), 23 to 29 nucleotide molecules that target transposons in the germline (Brennecke et al. 2007) and also function in somatic tissues, including the brain (Wakisaka and Imai 2019; Kim 2019). Transposons are mobile genetic elements which promote genetic diversity; however, if left unchecked to insert in the genome, will result in hybrid dysgenesis and mutations in flies (reviewed in (McCullers and Steiniger 2017)). The biogenesis of piRNAs falls into two categories: the production of primary piRNAs, whereby a piRNA precursor is transcribed from the genome and processed into mature piRNAs, and the production of secondary piRNAs using a primary piRNA as a template (Figure 1B) (Czech and Hannon 2016). Secondary piRNAs act post-transcriptionally to silence active transposons (Czech and Hannon 2016).
Transposon expression and activity increase during aging in flies (Li et al. 2013), and as piRNAs have been shown to target transposons to reduce their activity, it is hypothesized that piRNAs play a protective role against transposon-induced genome damage during aging. While studies investigating piRNAs in the circadian system are sparse, previous work in our lab has shown that five putative piRNA-containing transcripts show both increased expression level and increased diurnal rhythmicity in the heads of aged flies (Kuintzle et al. 2017).
Although many Drosophila small RNAs are well-studied as individual regulators in young animals, their systems-level role in circadian or light-activated regulation of transcript expression during aging is not understood. To assess the relationship between diurnal small RNA expression and the aging circadian clock output, we performed small RNA sequencing around-the-clock on the heads of young (5 day) and old (55 day) female D. melanogaster. In this work we analyzed age-related expression and rhythmicity changes in microRNAs and piRNAs, and investigated the potential biological effects of microRNA expression changes with age by integrating this small RNA dataset with an RNA-seq dataset previously published by our lab (Kuintzle et al. 2017). In addition, we identified novel age-onset microRNAs and transfer-RNA derived fragments (tRFs) and characterized changes in their expression during aging.
The animals used in this study do not require ethics approval in accordance with Oregon State University’s Institutional Animal Care and Use Committee.
Mated female Drosophila melanogaster (w1118) were mainatined at 25°C on a standard agar (7.4g/L) (Genesee Scientific, Catalog number: 66-103), cornmeal (50g/L) (Genesee Scientific, Catalog number: 62-101), yeast (35g/L) (Genesee Scientific, Catalog number: 62-103), and molasses (5%) (Genesee Scientific, Catalog number: 62-117) diet under a light-dark (LD) 12h:12h cycle and housed in groups of 50 flies in 300 ml round bottom polypropylene ventilated bottles (Genesee Scientific, Catalog number: 32-129F). Diet was changed without anesthesia three times a week. Two biological replicates of 400 flies were collected for young and old at 4 hour intervals around the clock. In parallel with samples for sequencing, 50 flies young and old were collected at each time point for potential verification of data by qPCR. At each time point, flies were transferred without anesthesia to Eppendorf tubes (Genesee Scientific, Catalog number: 22-282) that were pre-frozen at -80°C. Whole flies were stored at -80°C. Heads were separated from bodies by vortexing tubes (Scientific Industries Vortex-Genie 2, Catalog number: SI-0236) and sifting them over stainless steel sieves with mesh opening sizes of 710 μm and 425 μm (custom sieves ordered from Tokyo Screen Co). Samples were kept frozen throughout the process with liquid nitrogen, and stored at -80°C until RNA isolation was performed.
Each sample of 400 heads was homogenized in TRIzol (Thermo Fisher, Catalog number: 15596026) with a handheld motorized pestle (motor: Kimble, Catalog number: 749540-0000; pestle: Kontes, Catalog number: KT-749521-1590) following the manufacturer’s instructions. Samples were treated with rDNAse I (Takara, Catalog number: 2270A) following the manufacturer’s instructions, and then extracted with a 1:1 ratio of sample volume tophenol/chloroform (Thermo Fisher, Catalog number: 15593031) volume. RNA was then precipitated with 200 proof ethanol (using 2.5x of RNA sample volume) (Koptec, Catalog number: 2716) and 0.3M pH 5.5 sodium acetate (using 0.1x of RNA sample volume) (Invitrogen, Catalog number: AM9740). Finally, the resulting RNA pellet was rinsed twice with 75% ethanol (Koptec, Catalog number: 2716) and dissolved in 50 μl nuclease-free water (Invitrogen, Catalog number: AM9939). The concentration and purity of total RNA was assessed with a NanoDrop NanoDrop® ND-1000 (Thermo Fisher). RNA was extracted in the same way from 50 heads per sample, except the final RNA pellet was dissolved in 20 μl nuclease-free water instead of 50 μl, and allocated for qPCR.
Drosophila small RNA sequencing experiments are known to be overwhelmed by a 30-nucleotide rRNA fragment, if the fragment is not depleted (Wickersheim and Blumenstiel 2013). Therefore, we size-separated total RNA from each sample using 15% denaturing polyacrylamide-urea gel (polyacrylamide: Bio-Rad, Catalog #1610144; urea: Thermo Fisher, Catalog # 15505035). The gel was pre-run in 0.5X TBE buffer at a constant current of 40 mA for 30 minutes, and then samples were loaded and run at a constant current of 30 mA for approximately 1 hour. The gel was stained for 15 minutes with SYBR™ Gold Nucleic Acid Gel Stain (Thermo Fisher; Catalog number: S11494) in 0.5X TBE buffer following the manufacturer’s instructions, then small RNA in the size range of 18 to 29 nucleotides were cut out and gel fragments were mixed with 1 μl of glycogen (Thermo Fisher, Catalog #9510) as a carrier. RNA was eluted overnight using 0.3M sodium acetate precipitation (Invitrogen, Catalog number: AM9740). RNA was then precipitated with 200 proof ethanol (using 3x of the sample volume) (Koptec, Catalog number: 2716). The resulting RNA pellet was rinsed twice with 75% ethanol (Koptec, Catalog number: 2716) and dissolved in 7 μl nuclease-free water (Invitrogen, Catalog number: AM9939). Full protocol details are available on protocols.io (DOI: https://dx.doi.org/10.17504/protocols.io.yxmvm2xpog3p/v1) (Fey et al. 2022).
Prior to library preparation, the quality and concentration of the size-selected small RNA was assessed with a Qubit 3.0 fluorometer (Thermo Fisher, Catalog number: Q33216) and the appropriate Qubit microRNA assay kit (Thermo Fisher, Catalog number: Q32880) according to the manufacturer’s instructions. To ensure complete depletion of the 30-nucleotide rRNA fragment, a DNA oligo with sequence complementarity to the fragment (Wickersheim and Blumenstiel 2013) was added to samples during the library preparation stage (1–2 μl of 10 μM oligo stock was added to each sample of 10–50 ng RNA in 5μl).
Libraries were prepared using TruSeq Small RNA Library Prep Kit (Illumina; Catalog number: RS-200-0012) following the manufacturer’s instructions. Briefly, adapters were ligated to the 3' and 5' ends of the sample and reverse transcription followed by amplification created cDNA constructs based on the small RNA ligated with 3' and 5' adapters. This step selectively enriches RNA fragments with adapter molecules on both ends. The amplified cDNA constructs were pooled, then gel purified. The final pooled library was quantified by Qubit fluorometer using the dsDNA HS Assay kit (Thermo Fisher; Catalog number: Q33230). For quality control analysis, 1 μl of resuspended construct was loaded on an Agilent Technologies 2100 Bioanalyzer using a DNA-specific chip. Library pools were quantified by qPCR using an ABI 7500 fast instrument and KAPA Biosystems Library Quantification kit (KAPA Biosystems; Catalog numer: KK4824). Libraries were run on an Illumina HiSeq 3000 instrument, 50bp single end with 12 libraries per lane.
Reads were quality filtered and sequencing adapters were trimmed using skewer (Jiang et al. 2014), with parameters set for a minimum quality score of 30 and a minimum read length of 18, using the adapter sequence “TGGAATTCTCGGGTGCCAAGG”. Reads were aligned to the Drosophila melanogaster genome (BDGP release 6.21/dm6) with Bowtie using the parameters “-l 18 –n 1 -e 50 -a -m 50 -S --best --strata”. The number of hits to the genome for each read was included by adding NH tags to the output SAM files.
We used a custom Python script to quantify reads by mapping to the miRBase version 22 genome annotation file (release 6) (Kozomara, Birgaoanu, and Griffiths-Jones 2019). Of the 495 microRNAs annotated in mirBase, we detected 458 in our experiment. We adjusted reads to account for paralogous microRNAs (which map to multiple places in the genome) by dividing each count by the number of hits to the genome, stored in the NH tags in the SAM file for each sample. Final quantification is in adjusted Reads per Million Mapped MicroRNAs (aRPMMM). We used similar custom Python scripts to quantify reads mapping to piRNAs.
Diurnal rhythmicity was computed using a Fourier-based method previously developed by our lab, whereby the relative power of the 24-hour period (RP24) is used as a measure of diurnal rhythmicity (Sebastian et al. 2022). We calculated an RP24 score for each microRNA using their around-the-clock expression profiles and calculated statistical significance for microRNAs with RP24 scores ≥ 0. MicroRNAs with q-values ≤ 0.05 were considered significantly rhythmic, and microRNAs with q-values > 0.05 were considered arrhythmic. Rhythmic transcripts were identified previously using the RP24 method (Sebastian et al. 2022).
We compared the global expression of small RNAs in young and old flies using a t-test, implemented with SciPy tools version 1.0.0 (Jones et al. 2001). All 12 samples in young flies comprised one group, and all 12 samples in old flies comprised another group. The means of the two groups were considered significantly different with a p-value ≤ 0.05.
We performed differential expression analysis using the programs edgeR (Robinson and Oshlack 2010) and DESeq2 (Love, Huber, and Anders 2014). We grouped all 12 replicates corresponding to young flies and all 12 replicates corresponding to old flies to identify microRNAs differentially expressed independent of time of day. MicroRNAs with a Benjamini-Hochberg adjusted p-value ≤ 0.05 were considered significantly differentially expressed.
We used the command-line tool TargetScan (TargetScanFly version 7.2) (Agarwal et al. 2018) to predict mRNA targets of all microRNAs. We filtered the predicted target transcripts by expression, including only those passing a threshold of 1 FPKM (Fragments per Kilobase per Million) in our around-the-clock dataset (Kuintzle et al. 2017). For the novel small RNA analysis, small RNAs with identical seed sequences were grouped into families, and TargetScan was used to predict target transcripts as described above.
We filtered DE microRNAs to include only those with a log2 fold change ≥ 0.58 (corresponding to a simple fold change of 1.5) and expression levels ≥ 10 aRPMMM. We filtered predicted target transcripts of DE microRNAs to include only those with expression levels ≥ 10 FPKM and a probability of conserved targeting score (PCT) ≥ 0.80. PCT is calculated by TargetScanFly to provide a measure of the conservation of a microRNA site in a target transcript sequence (Friedman et al. 2009). We performed pathway analysis using the DAVID functional clustering and annotation webtool (DAVID 2021 update) with default parameters (Huang et al. 2007; Huang da, Sherman, and Lempicki 2009).
To determine if predicted microRNA target transcripts show evidence of post-transcriptional regulation, we performed exon intron split analysis (EISA) (Gaidatzis et al. 2015), examining the ratio of changes in exonic reads to changes in intronic reads between young and old flies for each transcript in our diurnal transcriptomic dataset (Kuintzle et al. 2017). We quantified intronic reads using bowtie to align FASTQ files to the Drosophila genome in an ungapped fashion using the parameters “--best --strata -m1”, then mapped the resulting reads to a FASTA file containing intronic reads downloaded from Flybase. We quantified exonic reads using bowtie to align FASTQ files to a FASTA file containing spliced transcript sequences using the parameters “-S --best --strata --all”, then mapped the resulting reads back to the spliced transcript FASTA file (also downloaded from Flybase). For each age, we used the sum of counts across all time points and replicates for exonic and intronic reads in this analysis.
We calculated an EISA score for each transcript in our dataset:
where Eold is exonic reads in old, Eyoung is exonic reads in young, Iold is intronic reads in old, and Iyoung is intronic reads in young flies. We calculated a two-tailed p-value for each transcript, performed multiple test correction to generate q-values for each transcript, and considered transcripts with q-values ≤ 0.05 to be statistically significant.We identified novel microRNAs using the random-forest-based novel microRNA identification program miRWoods (Bell et al. 2019). For each age, we removed novel microRNA primary transcripts with only one predicted product (keeping only primary transcripts with both 5' and 3' products), and retained only the mature products (5' and 3' arms) with median expression ≥ 1 aRPMMM. The GFF files from miRWoods output were used for quantification of novel microRNAs and are included in the Underlying data, Supplementary Files 11 and 12.
RNA extracted from young and old flies collected at different time points was used for reverse transcription of each sample. cDNA synthesis was achieved with the TaqMan MicroRNA Reverse Transcription Kit (Thermo Fisher, Catalog number: 4366596), used along with TaqMan Fast Advanced Master Mix (Thermo Fisher, Catalog number: 4444556) and the TaqMan™ microRNA assay (Thermo Fisher, Catalog number: 4440886) specific to miR-193-5p (Thermo Fisher assay ID 463359) and 2S rRNA (Thermo Fisher assay ID 001766), used as housekeeping reference gene.
For each biological replicate, equal amounts of cDNA from each time point were mixed to produce samples that were averaged across time points.
Real-time PCR was performed with Power SYBR Green PCR Master Mix (Thermo Fisher, Catalog number: 4367659) on a QuantStudio 3 Real-Time qPCR System (Applied Biosystems) according to the manufacter’s instructions for the TaqMan small RNA assays. Relative expression was calculated with the 2-ΔΔCT method, using 2S rRNA as the reference small RNA. Expression of 2S rRNA was compared across time points and ages in both the sequencing and qPCR analyses and determined to be the best candidate for a reference small RNA, due to its constant expression across both time of day and age.
MicroRNA expression plots were produced using a custom Python script (Fey 2022). Expression plots may be generated and downloaded using this webtool.
Flies were collected at 5 days (young) or 55 days (old) of age, around-the-clock every 4 hours beginning at lights-on at Zeitgeber time 0 (ZT0). For each sample, 400 heads were pooled and extracted RNA was submitted for sequencing. Two biological replicates were used for each time point in both young and old ages.
We aligned processed reads to the Drosophila genome and quantified microRNA expression by counting reads overlapping mature product annotations from the miRBase version 22 GFF file (Kozomara, Birgaoanu, and Griffiths-Jones 2019). Reads were normalized relative to each million reads mapping to microRNAs, and expression values are reported in adjusted Reads per Million Mapped MicroRNAs (aRPMMM). We quantified piRNA expression at the cluster level by mapping reads to the cluster reference from the piRNA database piRNAdb version 1.7.6 and normalized relative to each million reads mapping to the genome (adjusted Reads per Million, aRPM). For novel small RNAs, reads were quantified by referencing the genomic coordinates in the GFF files produced by miRWoods (Bell et al. 2019) and normalized relative to each million reads mapping to the genome (adjusted Reads per Million, aRPM). MicroRNA expression values are available in the Underlying data, Supplementary File 1 (Hendrix and Fey 2022).
Gene expression profiles exhibiting diurnal rhythmicity are commonly used to identify genes regulated by the circadian clock, or directly or indirectly activated by light. We detected rhythmic microRNAs in this dataset using the RP24 method, a Fourier-based approach previously developed by our lab that measures the relative power of the 24-hour period compared to all other periods (Sebastian et al. 2022). A more positive score indicates a higher level of 24-hour rhythmicity. Briefly, we calculated the RP24 score for each microRNA passing an expression threshold of 1 aRPMMM, and considered microRNAs with a q-value ≤ 0.05 to be significantly rhythmic.
Our previous work found a statistically significant net increase in transcript rhythmicity with age by comparing transcript RP24 distributions between young and old flies (Sebastian et al. 2022). To examine net changes in microRNA rhythmicity with age, we compared microRNA RP24 value distributions in young and old flies. In contrast to our previous findings for transcript rhythmicity, there was no statistically significant difference in microRNA RP24 distributions between young and old flies, suggesting that net microRNA rhythmicity is unchanged during aging (Underlying data, Supplementary Figure 1). In addition, we directly compared transcript and microRNA RP24 distributions for young and old flies and found that transcripts are significantly more rhythmic than microRNAs in both ages (Figure 2A).
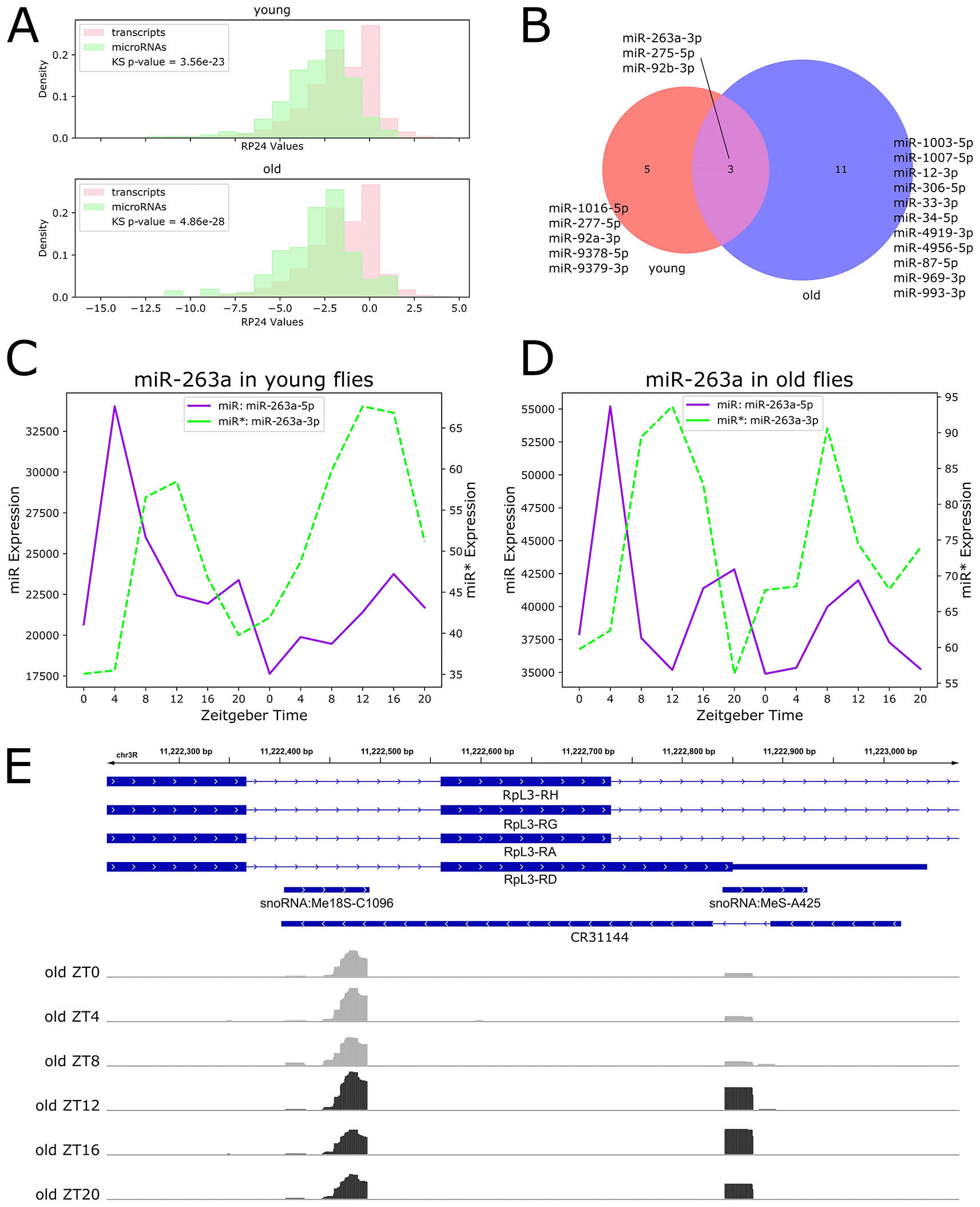
We next investigated individual microRNAs with significant diurnal rhythmicity. To characterize age-related changes in rhythmicity, we grouped rhythmic microRNAs into three categories: 1) microRNAs rhythmic in young flies and arrhythmic in old flies, 2) microRNAs arrhythmic in young flies and rhythmic in old flies, and 3) microRNAs rhythmic in both ages. We identified only 19 rhythmic microRNAs: five that lost rhythmicity with age, and 11 that gained rhythmicity with age, and three that maintained rhythmicity throughout the lifespan (Figure 2B).
Among the five microRNAs that are rhythmic in young flies and arrhythmic in old flies were miR-277-5p, which is involved in branched chain amino acid catabolism and lifespan determination (Esslinger et al. 2013), and miR-92a-3p, which has been shown to cycle in PDF neurons (Chen and Rosbash 2017). Expression profiles for these examples are shown in the Underlying data, Supplementary Figure 2.
The three microRNAs rhythmic in both young and old flies were miR-92b-3p, miR-263a-3p, and miR-275-5p. Of these, miR-263a-3p, also known as bereft (bft), has previously been shown to be rhythmic and under the control of the circadian clock in young flies (Yang et al. 2008). Expression of these three examples are shown in the Underlying data, Supplementary Figure 3.
MicroRNAs that gain rhythmicity in old flies included miR-33-3p and miR-34-5p, which has previously been shown to be strongly upregulated with age in Drosophila brains (Liu et al. 2012). Expression profiles of these examples are shown in the Underlying data, Supplementary Figure 4.
Surprisingly, we found no microRNAs with statistically significant rhythmicity for both 5' and 3' products. Based on the microRNA transcription process (Figure 1A), rhythmic transcription of the microRNA primary transcript should result in rhythmic expression for both mature products; however, it has been shown that while the non-functional miR* is degraded in the cell, the functional miR is stabilized by association with Ago1 (Van den Brande et al. 2018). We hypothesized that incorporation into the RISC and subsequent stabilization may mask temporal expression patterns for functional miRs, which remain visible for the non-stabilized miR*. To test this hypothesis, we used relative expression levels to categorize rhythmic microRNAs as highly-expressed functional miRs or lowly-expressed miR* products. We found that eight out of the 19 rhythmic microRNAs are miR* products: miR-277-5p, miR-275-5p, miR-1003-5p, miR-12-3p, miR-1007-5p, miR-33-3p, miR-87-5p and miR-263a-3p. We visualized the expression of the rhythmic miR* miR-263-3p and the corresponding non-rhythmic miR-263a-5p in both young (Figure 2C) and old (Figure 2D) flies. We suggest examination of such miR* expression profiles may offer insights into circadian or light-activated microRNA regulation which is invisible in functional miR expression profiles.
We also identified diurnally-expressed piRNAs using the RP24 method and found only one significantly rhythmic cluster, which was arrhythmic in young flies and rhythmic in old flies (Underlying data, Supplementary Figure 5). We visualized this piRNA cluster on the Broad Institute’s Integrative Genomic Viewer (IGV) tool (Figure 2E) (Thorvaldsdottir, Robinson, and Mesirov 2013), and found that this cluster overlaps RpL3, encoding a ribosomal protein, as well as three noncoding RNAs: two small nucleolar RNAs hosted in RpL3, and the antisense RNA CR31144 located on the opposite strand. The full lists of rhythmic microRNAs are available in the Underlying data, Supplementary File 2.
Because we detected relatively few rhythmic microRNAs and piRNA clusters, we deemed it important to conduct age-dependent but time-of-day-independent analyses. To investigate global changes in small RNA expression with age, we examined the number of reads mapped to annotated microRNAs at all time points, separately for young and old flies, normalized relative to the total number of reads mapped to the genome (Figure 3A). We find that the number of microRNA reads is significantly lower in old flies (p-value < 0.001, t-test), indicating that global microRNA expression decreases with age.
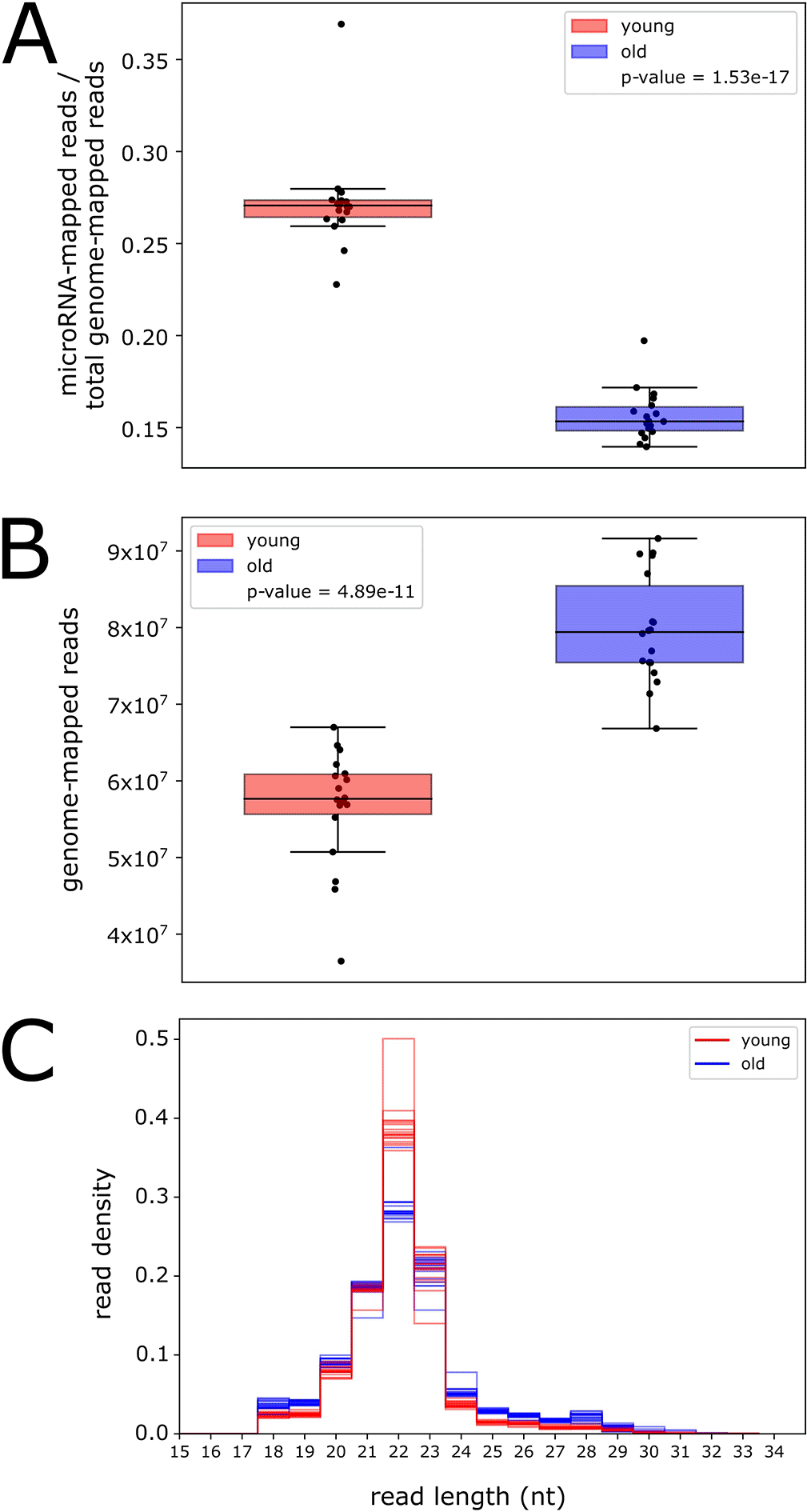
Global total RNA expression decreases during aging in Drosophila (Davie et al. 2018); therefore, we tested whether the observed global decrease in microRNA expression was due simply to a lower number of genome-mapped reads in old flies. Surprisingly, examination of the total number of reads mapped to the genome in both ages revealed a significantly higher number in old flies (p-value < 0.001, t-test, Figure 3B). This suggests that there is an upregulation in old flies of an RNA product with a similar size range as microRNAs. The read length distribution for all samples in young flies shows a strong, sharp peak at 22 nucleotides (nt) (Figure 3C), corresponding to the size of Drosophila microRNAs (Xue and Zhang 2018). In old flies, this 22-nt peak is severely dampened; however, there is an increase in genome-mapped reads from 24 to 29 nt, corresponding to the size range of piRNAs (Brennecke et al. 2007).
We hypothesized that the increase in genome-mapped reads in old flies is due to an age-associated upregulation in the expression of piRNAs, small RNAs that target transposons. Transposon expression and activity is known to increase during aging in fly heads (Li et al. 2013); therefore, piRNA expression may also increase as a protective measure. To test this hypothesis, we quantified piRNA reads and examined the number of reads mapped to piRNA clusters for young and old flies, normalized relative to the total number of genome-mapped reads (Underlying data, Supplementary Figure 6). We find that more reads map sense to piRNA clusters in old flies compared to young flies, suggesting an increase in global piRNA expression during aging. Interestingly, we also find an increase in reads mapping antisense to piRNAs. These reads may be derived from transposons, which are used as templates to produce piRNAs (Czech and Hannon 2016), suggesting that transposon expression may be increased in old flies in this dataset.
Our previous work identified five putative piRNA primary transcripts with age-induced rhythmicity (Kuintzle et al. 2017). We confirmed expression of small RNA reads deriving from these five primary transcripts in this dataset, with two expressed at levels ≥ 1 aRPM in at least one age (Underlying data, Supplementary Figure 7). The piRNA expression data and primary transcript genomic coordinates are available in the Underlying data, Supplementary Files 3 and 4.
To understand how the expression of individual microRNAs is affected in the heads of aging flies, we performed differential expression analysis using two R packages, DESeq2 (Love, Huber, and Anders 2014) and edgeR (Robinson and Oshlack 2010). We defined a high-confidence set of 170 differentially expressed (DE) microRNAs which were identified as significantly DE by both programs (Underlying data, Supplementary Figure 8). We found that 68 microRNAs were significantly upregulated and 102 were significantly downregulated in old flies compared to young.
We examined DE microRNAs which passed a median expression threshold of 1 aRPMMM in either age in Figure 4A, where labeled microRNAs have expression ≥ 5 aRPMMM. The most highly upregulated microRNA was the miR-2 family member miR-13b-2-5p, with the miR-2 family member miR-13a-5p also significantly upregulated with age. The microRNA miR-308-3p, which targets Myc, an ortholog to a human oncogene controlling cell growth and proliferation (Daneshvar et al. 2013), was also significantly higher in old flies compared to young.
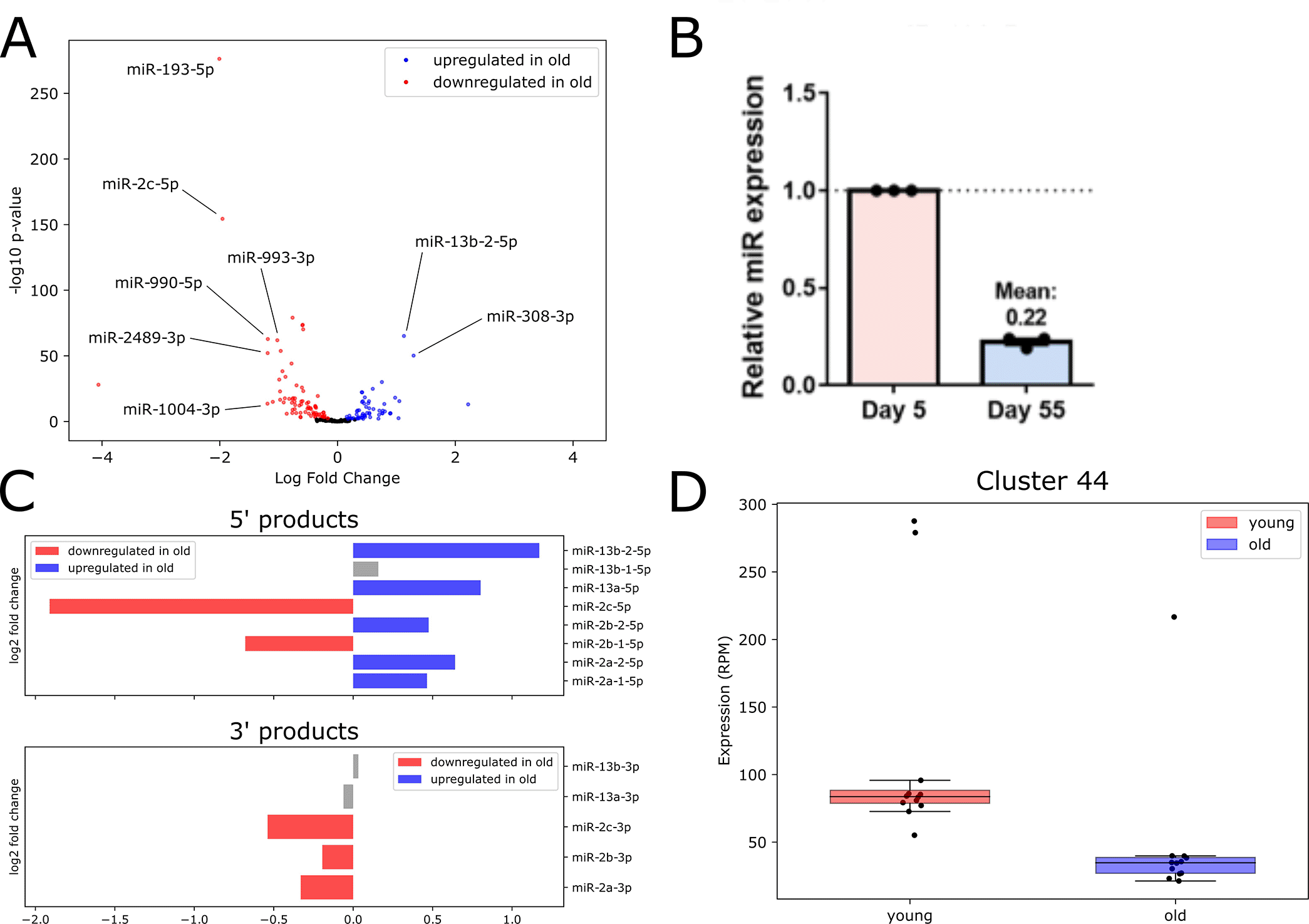
The most strongly downregulated microRNA in old flies compared to young was miR-193-5p, which has been implicated in the immune response in young flies (Atilano et al. 2017). We confirmed dramatic decrease in expression levels for miR-193-5p using RT-qPCR (Figure 4B). Also downregulated with age were miR-993-3p and miR-990-5p, the latter of which acts in glial cells to regulate circadian locomotor activity (You et al. 2018). Another member of the large multi-cluster miR-2 family, miR-2c-5p, was also downregulated in old flies.
The differential expression of various miR-2 microRNAs prompted us to investigate this family further. The miR-2 family of microRNAs is well-conserved in invertebrates: the 3' arm of microRNAs belonging to this family is very highly conserved, and is generally acknowledged as the functional miR, while the 5' miR* arm is more variable in sequence (Marco, Hooks, and Griffiths-Jones 2012). We observed no upregulation of 3' products; however, five 5' products were upregulated in old flies (Figure 4C). We also note a surprising inconsistency in expression changes with age for three clustered miR-2 family members (miR-2c, miR-13a and miR-13b-1), which are hosted in the long non-coding RNA (lncRNA) CR45911 and are thought to be under common transcriptional control. Both miR-2c-3p and miR-2c-5p are downregulated, while miR-13a-5p is upregulated in old flies. The remaining members of this cluster, miR-13a-3p, miR-13b-3p and miR-13b-1-5p, were not significantly differentially expressed in our data (Figure 4C). This difference in age-related expression changes among clustered miR-2 family members hints at an age-onset change in stability or processing for individual microRNAs.
We also performed differential expression analysis to identify age-dependent changes in piRNA cluster expression. We defined a high-confidence set of DE piRNAs which were detected with both edgeR (Robinson and Oshlack 2010) and DESeq2 (Love, Huber, and Anders 2014). We found two piRNA clusters significantly upregulated and two significantly downregulated with age (Underlying data, Supplementary Figure 9). Notably, the downregulated piRNAdb Cluster 44 (Figure 4D), which contains 16 individual piRNAs, overlaps the well-studied flamenco locus. The flamenco lncRNA hosts the largest piRNA cluster in Drosophila somatic cells, and acts to repress gypsy transposon activity (Ozata et al. 2019). This age-dependent downregulation of a proven piRNA-producing locus may partially explain observed increases in gypsy transposon expression and activity with age (Li et al. 2013). Full lists of DE small RNAs identified by each DE analysis program are available in the Underlying data, Supplementary Files 5 through 8.
To explore the potential biological impacts of the most highly DE microRNAs in our dataset, we first integrated a transcriptomics dataset previously generated by our lab (Kuintzle et al. 2017), then performed pathway analysis on the predicted targets of DE microRNAs. We filtered DE microRNAs to include those with absolute log2 fold change values ≥ 0.58 (corresponding to a simple fold change of 1.5) and expression values ≥ 10 aRPMMM, resulting in 21 downregulated and 13 upregulated microRNAs. We used the target prediction tool TargetScanFly version 7.2 (Agarwal et al. 2018) to predict target transcripts of each DE microRNA, then filtered the transcripts by expression and probability of conserved targeting score (see Methods). With these filters, downregulated microRNAs had 76 unique predicted target transcripts, and upregulated microRNAs had 174 unique predicted target transcripts. We used the DAVID functional annotation webtool (Huang et al. 2007; Huang da, Sherman, and Lempicki 2009) to identify functional pathways that may undergo changes in post-transcriptional regulation with age due to the differential expression of microRNAs.
We visualized the log2 fold change values for transcripts in each significant cluster resulting from analysis of predicted targets of upregulated (Figure 5A) and downregulated (Figure 5B) microRNAs. We noted that the transcripts belonging to these enriched pathways have low to moderate log2 fold change values. In some cases, transcript expression is altered in the same direction as the expression of the DE microRNAs predicted to target them. While this may seem counterintuitive at first glance, post-transcriptional regulation functions in concert with other determinants of expression level, such as basal transcription rate; therefore, change in expression level alone cannot be the sole evidence for microRNA targeting. In addition, it has been found that most microRNAs cause only modest changes in target expression, and in a system-wide context microRNAs are thought to act as buffers that protect against large changes in transcript expression for important pathways (Bartel 2018; Ebert and Sharp 2012). Therefore, targets of DE microRNAs may be post-transcriptionally regulated even though a large change in transcript expression with age is not observed.
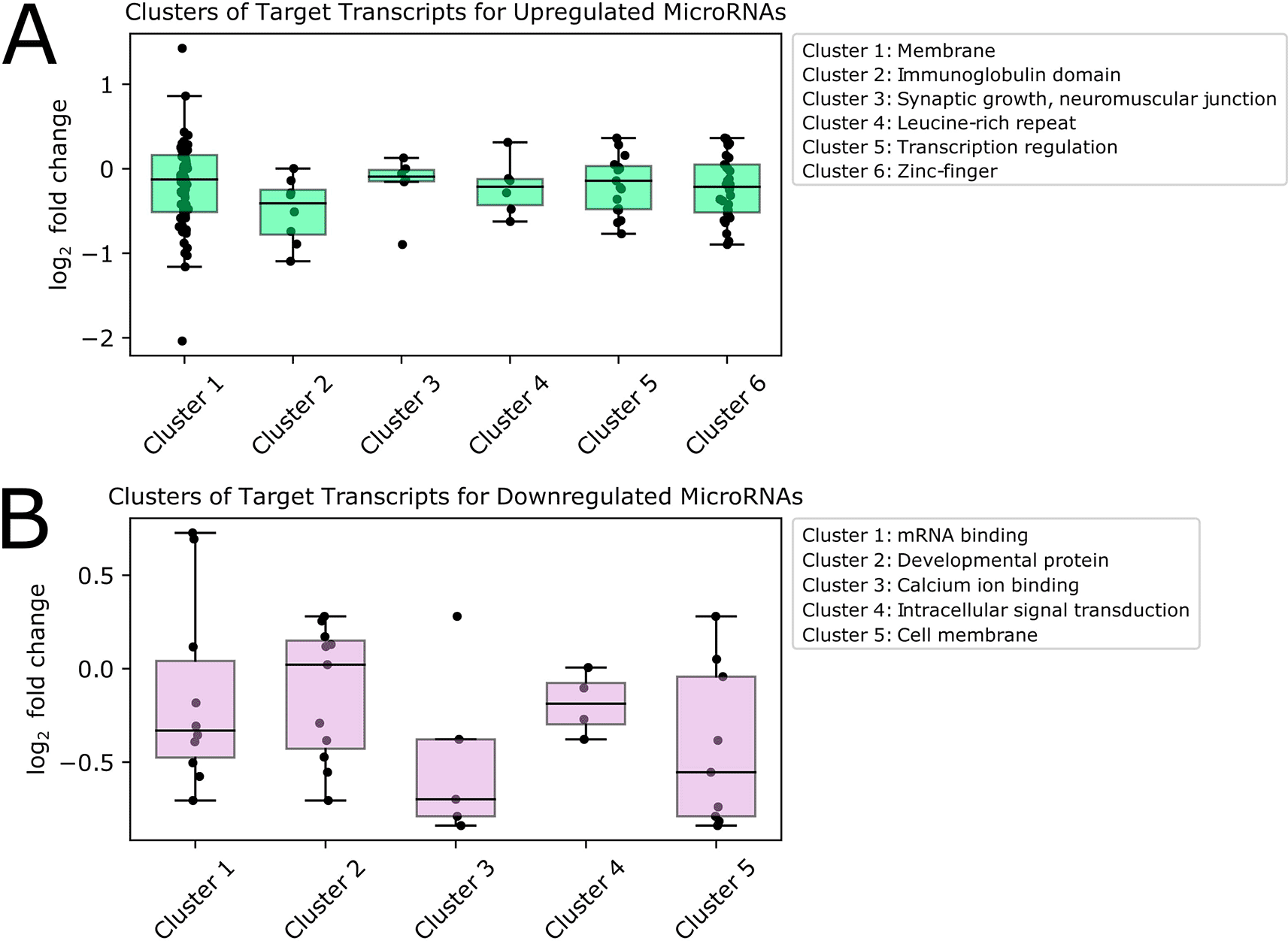
We investigated pathways associated with targets of upregulated microRNAs (Figure 5A) and found that the largest and most highly enriched pathway contained transcripts encoding membrane proteins, including Rh7-RA, a rhodopsin photoreceptor playing a role in circadian entrainment to light, and Nlg3-RC, a neuroligin involved in synaptic transmission. Predicted target transcripts also encoded transcription factors, including several related to stress and aging. Among these were Mnn1-RC, which regulates multiple stress response pathways, including hypoxia and oxidative stress, and REPTOR-RB (Repressed by TOR), which is involved in regulating stress and aging with TOR (target of rapamycin). Other predicted targets of upregulated microRNAs were involved in synaptic growth at the neuromuscular junction, and encoded proteins containing immunoglobulin, zinc finger, and leucine-rich repeat domains.
We also explored pathways associated with predicted targets of downregulated microRNAs (Figure 5B). We found that the most highly enriched group of transcripts encoded mRNA binding proteins, including elav-RD, which is neuronally expressed, and msi-RF, which encodes a protein that represses the hypoxia inducible factor (HIF) pathway by targeting the 3' UTRs of mRNAs. One of the genes targeted by Msi is the transcription factor ttk (tramtrack), which has been proposed as a putative regulator of pdf, the main circadian neuropeptide in Drosophila (Mezan et al. 2016). Also included in this group was mbf1-RB, which encodes a transcriptional co-activator that induces transcription of stress response genes, and has one of the highest log2 fold changes of transcripts in this cluster. Other transcripts predicted to be targets of significantly downregulated microRNAs were involved in calcium-ion binding and intracellular signal transduction, and encoded membrane and developmental proteins. Full pathway analysis results are available in the Underlying data, Supplementary File 9.
Next, we refined our exploration of the potential biological effects of age-related microRNA expression changes on predicted target transcripts. We used a previously published method, exon intron split analysis (EISA), to determine the extent to which a transcript is likely undergoing changes in post-transcriptional regulation with age (Gaidatzis et al. 2015). For every transcript we calculated an EISA score:
where Eold and Eyoung are the sum of reads across time points mapping to exons in old and young flies, respectively. Similarly, Iold and Iyoung are the sum of reads across time points mapping to introns in old and young flies, respectively. The final score compares reads mapping to transcript exons and introns between young and old conditions. Transcripts with a negative EISA score have a higher proportion of intronic reads compared to exonic reads, and are therefore likely experiencing increased microRNA targeting after aging. We calculated a q-value for each transcript, and retained those with a q-value ≤ 0.05 and a median expression ≥ 1 FPKM (Fragments per Kilobase per Million) in either age.We found that 98 transcripts had a significant EISA score, indicating that they are likely experiencing age-related changes in post-transcriptional regulation (Figure 6A). We found 38 transcripts with a positive score, and 60 with a negative score, suggesting that over one-and-a-half times as many transcripts experience increased rather than decreased post-transcriptional regulation in old flies according to this score. We noted that many of these transcripts do not show strong expression differences between young and old flies, highlighting the usefulness of this score for uncovering evidence of post-transcriptional regulatory changes with age.
We next investigated which microRNAs might be responsible for mediating these changes in post-transcriptional regulation with age. We focused our analysis on significant transcripts that have predicted binding sites for DE microRNAs that pass a median expression threshold of 1 aRPMMM in either age. Next, we ensured that the calculated transcript EISA score was anti-correlated with the log2 fold change of the DE microRNA; that is, we kept transcripts with negative EISA scores predicted to be targeted by upregulated microRNAs, and transcripts with positive EISA score predicted to be targeted by downregulated microRNAs (pairs available in the Underlying data, Supplementary File 10). We found that 79 significant transcripts had predicted binding sites for DE microRNAs with anti-correlated log2 fold change values (Underlying data, Supplementary Figure 10).
We examined the strongest microRNA-transcript pairs: microRNAs with absolute log2 fold change values ≥ 0.58 (corresponding to a simple fold change of 1.5) and predicted targets with EISA scores ≥ 1.5 or ≤ -1.5. We found 19 transcripts corresponding to 15 genes passing these thresholds. Interestingly, 10 of these transcripts have demonstrated roles in the maintenance or regulation of muscle.
We first examined transcripts showing evidence of increased post-transcriptional regulation with age (Figure 6B). The transcript in this group with the strongest EISA score was Boot-RA, which is involved in the nuclear export of piRNAs (ElMaghraby et al. 2019). We also found eight transcripts known to be involved in muscle function: SERCA-RA, Mlp60A-RE, Mlp84B-RA, Mlp84B-RB, Mlp84B-RC, Prm-RE, Act-87E-RA, and Nlg1-RD. SERCA-RA encodes an endoplasmic reticulum calcium pump, and Mlp60A-RE encodes a muscle LIM-domain protein which maintains flight muscles; both have been implicated in the molecular mechanism underlying aging muscle (Bordet et al. 2021; Delrio-Lorenzo et al. 2020). Mlp84B is another muscle LIM protein with roles in muscle maintenance (Clark, Bland, and Beckerle 2007). An additional transcript in this group was Nlg1-RD (Neuroligin 1), encoding an adhesion protein that plays a role in the function of postsynaptic neuromuscular junctions (Gaudet et al. 2011; Banerjee, Venkatesan, and Bhat 2017; Owald et al. 2012). Lastly, we found Prm-RE, encoding an invertebrate-specific muscle protein (Becker et al. 1992), and Act87E-RA, an actin with diverse roles, including in muscle contraction (Roper, Mao, and Brown 2005).
We found that many of these muscle-related transcripts are predicted targets of a small subset of microRNAs, including miR-9a-5p, which plays diverse roles in development, including regulating the formation of the junction between muscle and tendons (Yatsenko and Shcherbata 2014). It is predicted to target five of these eight transcripts. Several of these microRNAs have been shown to be expressed in Drosophila thoracic muscle (Fulga et al. 2015), including the circadian-associated let-7-5p, which is predicted to target five of these transcripts, and the circadian-regulated miR-263a-5p, which is predicted to target Act87E-RA and Nlg-RD. Mlp84B-RC and Act87E-RA are predicted targets of miR-13a-5p, which is also expressed in thoracic muscle. Expression of a miR-13a sponge in thoracic muscle results in flies with impaired flight phenotypes (Fulga et al. 2015).
We also examined transcripts showing evidence of decreased post-transcriptional regulation with age (Figure 6C). These included Tm2-RB and Tm2-RG, encoding tropomyosin 2, which is critical for regulating calcium-dependent muscle contraction. Both isoforms have predicted binding sites for downregulated miR-133-5p, a conserved microRNA that regulates muscle development in mice and Xenopus (Boutz et al. 2007; Chen et al. 2006). This group also included Rnmt-RA, which encodes a methyltransferase that caps mRNAs. Taken together, these results suggest the altered expression of microRNAs potentially involved in muscle-regulatory networks during aging.
We used the random-forest-based microRNA detection program miRWoods (Bell et al. 2019) with the goal of identifying novel microRNAs in our dataset. We ran the program separately on data from young and old flies, and identified 121 novel small RNA primary transcripts between the two ages. We retained primary transcripts which had two predicted mature products (5' and 3' arms), resulting in a high-confidence set of 58 novel small RNA primary transcripts. We filtered the corresponding mature products by expression level and retained 24 novel mature small RNA products: four were detected only in young flies, eight were detected in both ages, and 12 were detected only in old flies (Figure 7A).
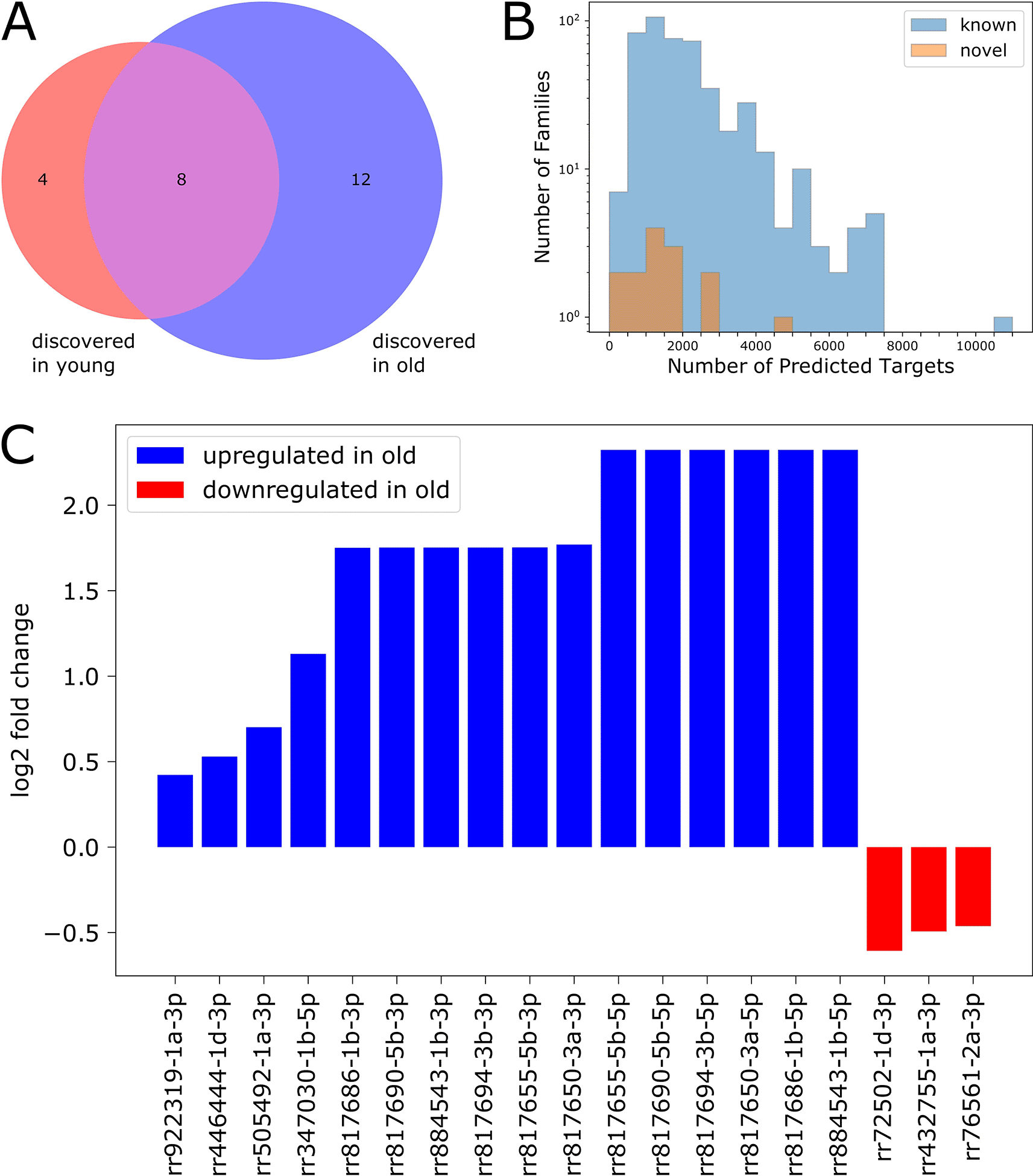
We used TargetScanFly (Agarwal et al. 2018) to predict target transcripts for each novel small RNA family (see Methods), then filtered the results to include only transcripts expressed at levels ≥ 1 FPKM in our dataset. We found that most of the newly identified small RNAs are predicted to target between 400 and 2000 transcripts, while many previously characterized microRNAs are predicted to target appreciably more transcripts (Figure 7B).
We performed differential expression analysis (as described above) to characterize age-associated expression changes for each novel discovery (Figure 7C). We found that 19 out of the 24 novel small RNAs were differentially expressed with age: 16 were upregulated in old flies, and three were downregulated in old flies.
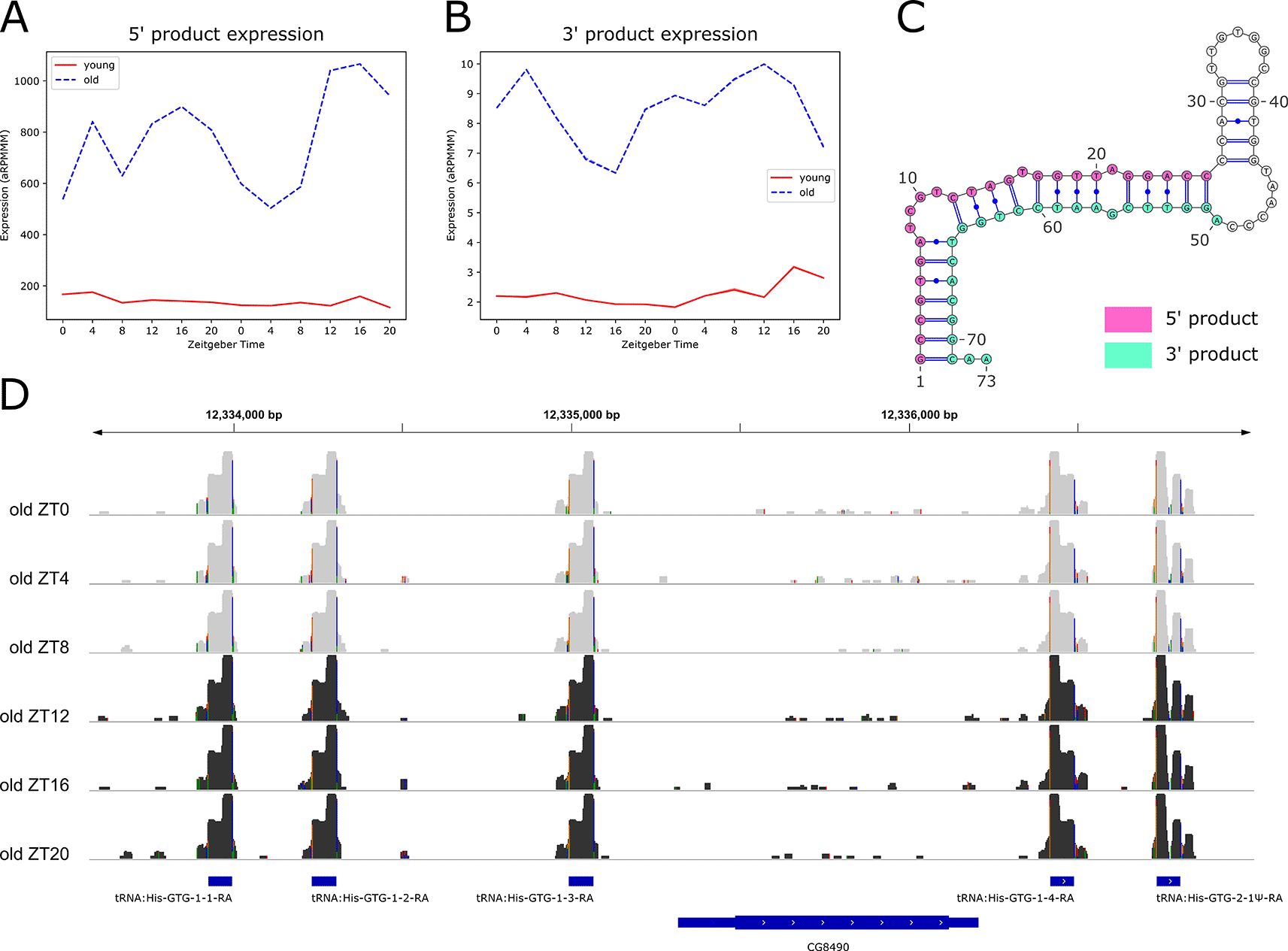
We also performed rhythmicity detection using the RP24 score to identify small RNAs with 24-hour periodicity that may be regulated by light or by the circadian clock. We found six rhythmic small RNAs, all of which were arrhythmic in young flies, but gained rhythmicity in old flies, with peak expression at ZT16 (Figure 8A). Notably, these six small RNAs identified as rhythmic in old flies were all identified as significantly upregulated with age.
Surprisingly, we found that these six rhythmic predicted microRNAs were novel transfer RNA derived fragments (tRFs). These recently discovered small molecules are hosted in mature or precursor tRNA sequences, and are classed according to the region of the tRNA from which they derive (Kumar et al. 2015; Krishna et al. 2021). Two classes map to mature tRNAs: tRF-5s map to the 5' end, and tRF-3s map to the 3' end of mature tRNAs. Those deriving from tRNA precursor sequences are named tRF-1s. Previous studies have observed tRFs in bacteria, plants, and animals, including Drosophila, mouse, and humans (Krishna et al. 2021).
We observed that the expression profiles for the six rhythmic tRFs overlap, and that they are all 5' products (Figure 8A). We examined expression profiles of their corresponding 3' products and found that these also overlap (Figure 8B), suggesting a high sequence similarity among 5' and among 3' products. Indeed, we found that all six of the 5' products share an identical 26-nt sequence, while the corresponding six 3' products share an identical 24-nt sequence. Further investigation revealed that the primary transcript sequences for five of these products are also identical, resulting in identical predicted dot-bracket structures, which we visualized using the RNA structure visualization tool VARNA (Figure 8C). The sequence of the sixth tRF primary transcript differs slightly, producing a different predicted structure (Underlying data, Supplementary Figure 11A). We conclude that these tRF loci exhibit microRNA-like characteristics, because miRWoods uses a random forest trained on known microRNA features, and we hypothesize that the predicted hairpin structure of the tRF primary transcripts contributed to the identification of these products with miRWoods (Bell et al. 2019).
We visualized the log-transformed reads for these tRFs using IGV (Figure 8D) and found that five of the six primary transcripts are clustered on either side of the uncharacterized gene CG8490. The sixth primary transcript is located on the same chromosome (2R), approximately 1000 base pairs downstream of the long noncoding RNA (lncRNA) CR44472. Visualization on the IGV tool confirmed that five of the primary transcripts map to loci encoding GTG Histidine transfer RNAs (tRNAs), and the sixth maps to tRNA:His-GTG-2-1Ψ-RA, a pseudo-tRNA (Figure 8D and Underling data, Supplementary Figure 11B).
The alignment patterns of these pairs of 5' and 3' products to tRNAs suggests that they are pairs of tRF-5 and tRF-3 molecules. We searched the tRF database tRFdb (Kumar et al. 2015) for these tRFs using genomic coordinates and sequences, and concluded that these are not currently annotated as tRFs. Thus, we report the discovery of novel tRFs which show age-onset expression and rhythmicity.
Here we present a systems-level exploration of diurnal changes in small RNA expression with age, including microRNAs, piRNAs, and the newly-discovered class of small RNAs, transfer RNA derived fragments (tRFs). We report an age-related increase in microRNA rhythmicity, but a global decrease in microRNA expression with age, corroborating results from studies in C. elegans, mice and humans (Ibanez-Ventoso et al. 2006; Inukai et al. 2012; Noren Hooten et al. 2010), and show a corresponding increase in global piRNA expression levels with age, suggesting a shift in the regulatory small RNA profile of Drosophila during aging.
An important contribution from our study is the identification of novel tRFs using the random-forest-based microRNA detection program miRWoods (Bell et al. 2019). Similar to microRNAs, tRFs have seed sequences which match transcript 3' UTR regions conserved in Drosophilids (Lee et al. 2009; Karaiskos et al. 2015). Additionally, they are loaded into Ago1 and Ago2 in Drosophila, and in some cases, they have been shown to silence target transcripts (Krishna et al. 2021; Karaiskos et al. 2015).
We found 12 novel tRFs (six tRF-5s and six tRF-3s) hosted in five tRNAs and one pseudo-tRNA. All 12 are upregulated in old flies, and the six tRF-5s are rhythmic in old flies but not in young, hinting at an age-associated regulatory role for these newly-discovered tRFs. While tRFs have been shown to be age-induced in Drosophila (Karaiskos et al. 2015), the characterization of 24-hour rhythmic expression patterns is a novel finding. Notably, we do not observe the canonical ‘CCA’ trinucleotide for the novel tRFs mapping to the 3' end of the mature tRNAs, suggesting that these tRF-3s derive from tRNAs which have not been fully processed into mature products. There is evidence for age-dependent changes in expression and activity of some tRNA modifying enzymes (Zhou et al. 2021); it is possible that changes in these modifications may alter the way in which tRNAs fold into their classic functional tertiary structure. Further, the program miRWoods identifies novel microRNAs based on the similarity of primary transcript sequences to a hairpin structure. This suggests that in the absence of chemical modifications required for proper folding, these tRNA molecules resemble a primary microRNA transcript. It is possible that age-related changes in tRNA chemical modifications affect their processing, allowing for the biogenesis and expression of tRFs in old flies.
We identified several differentially expressed microRNAs which have previously been shown to have neuroprotective roles in aging Drosophila. These include the downregulated miR-1000-5p, which plays a neuroprotective role in light-dependent regulation of presynaptic glutamate release (Verma et al. 2015), and the upregulated miR-263a-3p, which similarly regulates glutamate excitotoxicity, specifically in glia (Aw et al. 2017).
Several microRNAs with predicted roles in immunity were downregulated with age in our data, including miR-1004-3p, which is predicted to target puc (puckered), a serine/threonine phosphatase involved in the Jun-N-terminal kinase (JNK) pathway (Fullaondo and Lee 2012), and miR-193-5p. The conserved miR-193-5p is involved in immune and stress response pathways in young flies and may play a role in buffering the response to infection (Atilano et al. 2017).
We found relatively few microRNAs with significant 24-hour periodicity, similar to previous microarray studies of diurnal microRNA expression in young flies (Yang et al. 2008; Vodala et al. 2012). Yang et al. showed miR-263a to be rhythmically expressed under the control of the circadian clock in young flies (Yang et al. 2008). This microRNA is part of the miR-263 family conserved in mammals, and has been shown to be expressed in Drosophila brains (Liu et al. 2012). We found that miR-263a-3p is upregulated with age, and we corroborate the study by Yang et al. showing that it is rhythmic in young flies. Further, we show that it maintains rhythmicity in old age.
We note that several miR* products are rhythmic in our dataset, while the corresponding functional miR is arrhythmic. Importantly, it has been shown that the functional miR is protected from degradation by binding to the stable Ago1 in Drosophila (Van den Brande et al. 2018). We propose a scenario in which the association of the functional miR with Ago1 may mask temporal expression patterns that are observable in the miR*. It has previously been shown that most circadian-regulated microRNAs in mouse liver are arrhythmic at the level of the mature microRNA product, but may be identified by the oscillations of the primary transcript (Wang et al. 2016). In a similar vein, we suggest that identification of oscillating miR* products may shed light on underlying regulation of microRNAs by light or by the circadian clock. We showed one example of a microRNAs with an arrhythmic functional miR and a rhythmic miR* products: the circadian-regulated miR-263a. Another example is miR-33-3p, which gains rhythmicity with age. When depleted in Drosophila glia, miR-33 affects rhythmic behavior, suggesting that it is involved in circadian regulation (You et al. 2018). Thus, we further suggest that age-related changes in circadian or light-activated regulation may be deduced by examining changes in miR* rhythmicity between young and old flies.
Another key finding of this study is the evidence for the age-induced increase in post-transcriptional regulation of transcripts involved in muscle maintenance and development. Our transcriptomic data are derived from whole heads, which contain several muscle groups operating the proboscis during feeding (McKellar et al. 2020). Several differentially expressed microRNAs are predicted to target these muscle-related transcripts. Transcripts showing evidence of decreased post-transcriptional regulation with age are predicted to be targets of the downregulated conserved miR-133-5p, which is muscle-associated in Xenopus and in mouse myoblasts (Chen et al. 2006; Boutz et al. 2007). The upregulated circadian-associated microRNAs let-7-5p and miR-263a-5p, both of which have been shown to be expressed in muscle tissue in Drosophila (Fulga et al. 2015), are predicted to target several transcripts that show evidence of increased post-transcriptional regulation with age. In addition, these transcripts are predicted targets of the upregulated miR-9a-5p, a regulator of muscle-tendon junction development (Yatsenko and Shcherbata 2014). Among these are all three isoforms of the gene Mlp84B, which has been shown to be downregulated with age in whole Drosophila (Bordet et al. 2021). Interestingly, we find that the most highly expressed isoform of Mlp84B, Mlp84B-RA, is upregulated in old flies in our transcriptomics dataset. This difference may stem from tissue-specific expression patterns for this transcript. We note that the use of whole fly heads prevents us from drawing definite conclusions about the potential for microRNA targeting of specific transcripts because we do not know that both the microRNA and its target are expressed in the same cell. However, these results hint at an age-onset reprogramming of muscle regulatory networks and suggests that a limited set of microRNAs may play a large role in regulating muscle during the aging process. We propose these microRNA-target pairs for future experimental analyses to confirm expression in the same cell and tissue types, prior to validation of targeting and biological impact.
The insights gained from this characterization of diurnal small RNAs in both young and old Drosophila advance our understanding of age-related changes in small regulatory molecule expression, and set the stage for continued exploration of the role of these players in aging circadian systems.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)