Keywords
single-cell RNA-seq, gene expression, workflow, Python, R
This article is included in the Cell & Molecular Biology gateway.
This article is included in the Bioinformatics gateway.
This article is included in the Python collection.
single-cell RNA-seq, gene expression, workflow, Python, R
Since a method for single-cell RNA sequencing (scRNA-seq) was first proposed (Tang et al., 2009), a number of diverse technologies have emerged for studying gene expression at single-cell resolution. Among these, droplet-based, 3′-end sequencing technologies such as Drop-seq (Macosko et al., 2015), inDrop (Klein et al., 2015), and in particular, 10X Genomics Chromium (Zheng et al., 2017) have become increasingly popular for studying gene expression across different cell types and cellular states due to their lower sequencing cost per cell, and higher throughput relative to other available technologies. These technologies rely on the use of short unique molecular identifier (UMI) sequences that are used to both quantify gene expression and reduce technical variation inherently present in scRNA-seq datasets (Islam et al., 2014; Kivioja et al., 2011). However, datasets derived using these UMI count-based technologies need to undergo several preprocessing and quality control steps before they can be used to address biological hypotheses.
As a result, hundreds of software tools have been developed to perform these preprocessing and quality control steps, with the vast majority of these software tools having historically been built for the R software environment (Zappia et al., 2018). One of the most popular R packages is undoubtedly Seurat (Stuart et al., 2019), which has become an integral component of many scRNA-seq workflows due to the wide range of analysis methods it offers and the utility of its custom data structures. However recently, an increasing number of new tools are being developed using the Python programming language (Zappia & Theis, 2021). One such tool is kb-python (Bray et al., 2016; Melsted et al., 2021), a wrapper for the kallisto | bustools scRNA-seq workflow (Melsted et al., 2021). Therefore there is a growing need to develop tools that enable integration of widely used existing R-based tools like Seurat and newer Python-based tools and analysis workflows like kb-python.
We therefore introduce SASCRiP (Sequencing Analysis of Single-Cell RNA in Python), a flexible and modular Python package designed to integrate kb-python, selected Seurat features, and additional custom data processing and visualization functions in a way that simplifies and streamlines preprocessing and quality control of UMI count-based scRNA-seq data in preparation for downstream analyses like clustering, data integration, and differential gene expression analysis.
SASCRiP (Moonsamy, 2021a) implements preprocessing of scRNA-seq datasets through four main functions, namely kallisto_bustools_count, seurat_matrix, run_cqc, and sctransform_normalize (Figure 1).

The SASCRiP workflow begins with the input of FASTQ files, and a Seurat object and mtx matrix containing gene expression values are produced as output. All SASCRiP functions, namely kallisto_bustools_count, seurat_matrix, run_cqc, and sctransform_normalize, produce intermediate files that are used as input into the following function.
The kallisto_bustools_count function wraps functions from the kb-python package, which itself wraps functions from the kallisto | bustools workflow. This function takes as input unprocessed paired-end FASTQ files derived from UMI-based scRNA-seq experiments. These are then, by default, pseudoaligned to a user-specified indexed transcriptome. The user also has the option to generate a new index and transcript-to-genes mapping file if these are not available. All aligned sequences are stored within a barcode UMI set (BUS) file that contains the barcode and UMI sequences, as well as the aligned transcripts. The kallisto_bustools_count function uses BUStools (Melsted et al., 2019) to remove polymerase chain reaction (PCR) duplicates and correct the barcode sequences that differ by 1 hamming distance from a barcode whitelist. The kallisto_bustools_count function includes barcode whitelists for 10x Genomics Chromium v1, v2 and v3, as well as for InDrops v3 data. For other UMI-based technologies, kallisto_bustools_count includes parameters that allow for the use of user-supplied barcode whitelists. By default, the BUS file is filtered to remove barcodes with no corresponding transcript information. Gene-level count matrices are then returned in the matrix market (mtx) format, where cells and genes are represented in rows and columns, respectively.
import sascrip from sascrip import sascrip_functions sascrip_functions.kallisto_bustools_count( list_of_fastq_files, # list of all the input FASTQ files in the required order single_cell_technology, # type of single-cell tech used to generate the data output_directory_path, # path where all output files will be saved species_index, # path to the kallisto index for the species of interest species_t2g # path to the transcripts-to-genes mapping file for the species of interest )
SASCRiP also includes an additional, optional function, edit_10xv1_fastq, that transforms scRNA-seq data obtained from experiments using 10x Genomics Chromium v1 technology into a format compatible with the kallisto | bustools workflow. Although these types of files are already supported by the kallisto | bustools workflow, most FASTQ files stored in public sequencing data repositories/databases are not provided in the required format. In situations where only two FASTQ files (one containing the transcript and UMI sequences, and the other containing the barcode sequences) are provided, the edit_10vx1_fastq function separates the UMI and transcript sequences into two new FastQ files, so that the three files required (containing the transcript, UMI, and barcode sequences) are available as input for the kallisto | bustools workflow.
import sascrip from sascrip import sascrip_functions sascrip_functions.edit_10xv1_fastq( directory_with_fastqs, # path to directory containing 10xv1 FastQ files output_directory # path where edited FastQ files will be stored )
Count matrices obtained using the kallisto | bustools workflow cannot be directly imported into Seurat. Therefore the SASCRiP function, seurat_matrix, converts the output obtained from BUStools (where genes are represented as rows and cells as columns) into a format compatible with Seurat, where cells are represented as rows and genes as columns. The seurat_matrix function also converts gene identifiers in the transcript-to-gene index file from the kallisto | bustools workflow from ENSEMBL IDs to their corresponding HGNC gene symbols, as required by Seurat.
import sascrip from sascrip import sascrip_functions sascrip_functions.seurat_matrix( mtx_bustools_matrix, # mtx matrix generated by kallisto_bustools_count bustools_gene_index, # genes index file generated with the mtx matrix bustools_barcode_index # barcode index file generated with the mtx matrix transcript_to_genes_file # path to transcript-to-genes files used/generated # with kallisto_bustools_count output_directory # path where Seurat-compatible files will be saved )
Gene-level count matrices obtained from either Seurat or SASCRiP’s seurat_matrix function can then be used as input for SASCRiP’s run_cqc function, which leverages Seurat’s custom data structures to identify low quality cells. The run_cqc function classifies cells as being of low quality based on two per-cell metrics: the total number of genes detected, and the percentage of sequencing reads that map to mitochondrial genes. These metrics are calculated automatically when a Seurat object is created. By default, run_cqc first removes cells with fewer than 200 genes (Ilicic et al., 2016), then cells where more than 10% of sequencing reads map to mitochondrial genes Osorio & Cai (2021). However, all parameters used to distinguish low quality cells from high quality cells can be defined by the user.
The run_cqc function also calculates the median absolute deviation (MAD) of the total number of genes per cell, allowing for the identification and removal of cell doublets. SASCRiP prioritises the removal of heterotypic doublets, such that, by default, cells with a total gene count higher than six MAD values from the median are classified as outliers and removed. The MAD values and outlier thresholds are calculated using the following equation:
Here, x represents the total gene count for all cells, xi is the gene count for a given cell, and n is the number of MAD values used to classify a cell as an outlier. The MAD value cut-off threshold can be manually defined by the user. Alternatively, other outlier detection methods, such as standard deviation, are also included within SASCRiP and can be selected as a substitute to MAD.
import sascrip from sascrip import sascrip_functions sascrip_functions.run_cqc( input_files, # directory containing Seurat-compatible files "sample", # user-given name of sample output_directory # path where all output files will be saved )
Multiple output files are produced by the run_cqc function. Cell quality control metrics and UMI count data are returned as Seurat objects, in rds format, and a log file is created. The cell quality control metrics can also be returned in tsv format, to facilitate data visualization; and UMI counts can be returned as an mtx matrix, for use with alternative analysis tools. In this way, output from SASCRiP can be easily integrated into alternative workflows and analysis pipelines that require different file formats.
SASCRiP performs normalization and variance stabilization through the sctransform_normalize function, which serves as a wrapper for sctransform (Hafemeister & Satija, 2019). In this way, technical variation present in the dataset due to differences in sequencing depth between cells is reduced. All parameters incorporated within the original sctransform::vst function can be modified through the sctransform_normalize function. Corrected UMI counts are log-normalized in order to obtain gene expression values and the 2000 most variable genes in the dataset are identified and returned. Normalized gene expression values or UMI counts may be returned, either as a Seurat object or as an mtx file.
import sascrip from sascrip import sascrip_functions sascrip_functions.sctransform_normalize( seurat_object, # path to saved subset Seurat object “sample”, # user-given name of sample output_directory # path where normalised data are saved )
The modularity of SASCRiP allows all functions to be implemented independently, but the package also includes an all-in-one function, sascrip_preprocess, that implements the entire workflow from start to finish. In order to ensure that appropriate quality control measures are applied at each stage of the workflow, quality control metrics are provided at multiple checkpoints in the SASCRiP workflow when the sascrip_preprocess function is executed. These metrics can be printed to the screen while running the workflow and/or written to file for later use.
All SASCRiP functions are designed and implemented in Python 3 (v3.7 or higher), and the package is available on PyPi. As SASCRiP also incorporates a number of R packages (including Seurat), R (v3.6 or higher) is also required. Any additional R packages required can then be installed through SASCRiP, if needed. SASCRiP was developed primarily for use on Unix-based operating systems, however the source code can be adapted for use on Windows platforms (as described in the SASCRiP documentation). SASCRiP can be used to process scRNA-seq datasets consisting of up to 10 000 cells on a standard laptop with 8 Gb RAM. At least 16 Gb of RAM is recommended for larger datasets.
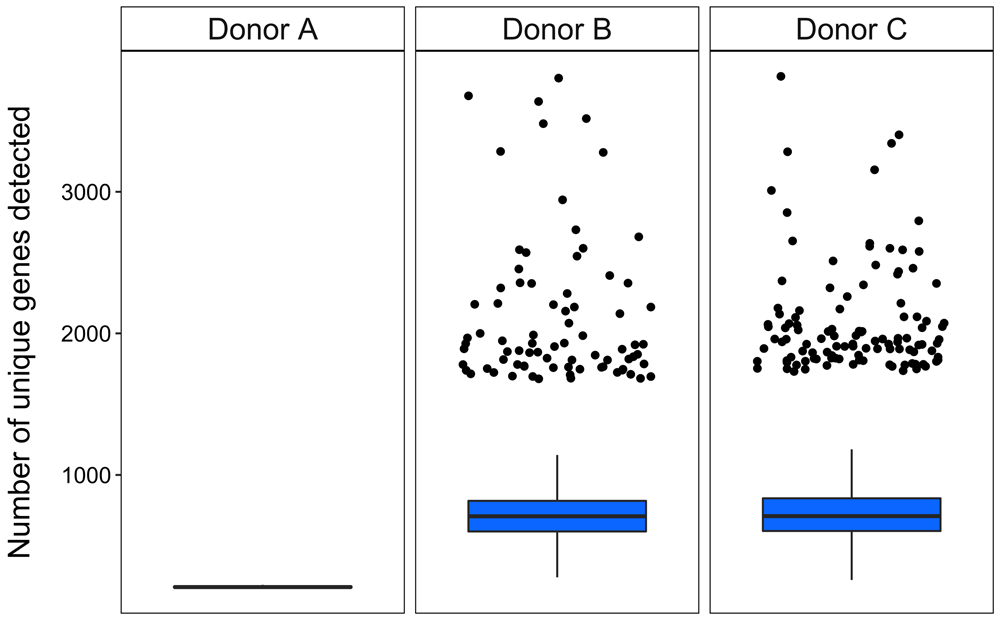
In order to demonstrate the utility of the SASCRiP workflow, we reanalyzed three scRNA-seq datasets derived from peripheral blood mononuclear cells (PBMCs) obtained from healthy individuals (Zheng et al., 2017). Cell counts range from 2000 to 10 000 cells per donor. SASCRiP’s sascrip_preprocess function was used to perform all preprocessing steps required to generate a high-quality gene expression dataset that could then be readily input into Seurat for cell clustering and annotation. All SASCRiP functions were implemented using the default parameters.
As these datasets were obtained using 10X Genomics Chromium v1 technology, SASCRiP’s edit_10xv1_fastq function was first used to generate the three FASTQ files required by the kallisto_bustools_count function. Gene-level count matrices were then obtained using the kallisto_bustools_count function, and cell quality control metrics were calculated using the run_cqc function (Moonsamy, 2021b). Visualization of these metrics allowed low quality cells (Figure 2) and cell doublets (Figure 3) to be easily identified and removed. These metrics, together with the UMI count data, were then input as Seurat objects, into the sctransform_normalize function. This allowed for normalization and variance stabilization of these UMI counts (Figure 4), so that gene expression could be quantified. In addition, the 2000 most highly variable genes, based on standardized variance, were identified (Figure 5). The normalization data was then output as a Seurat object, in preparation for clustering.
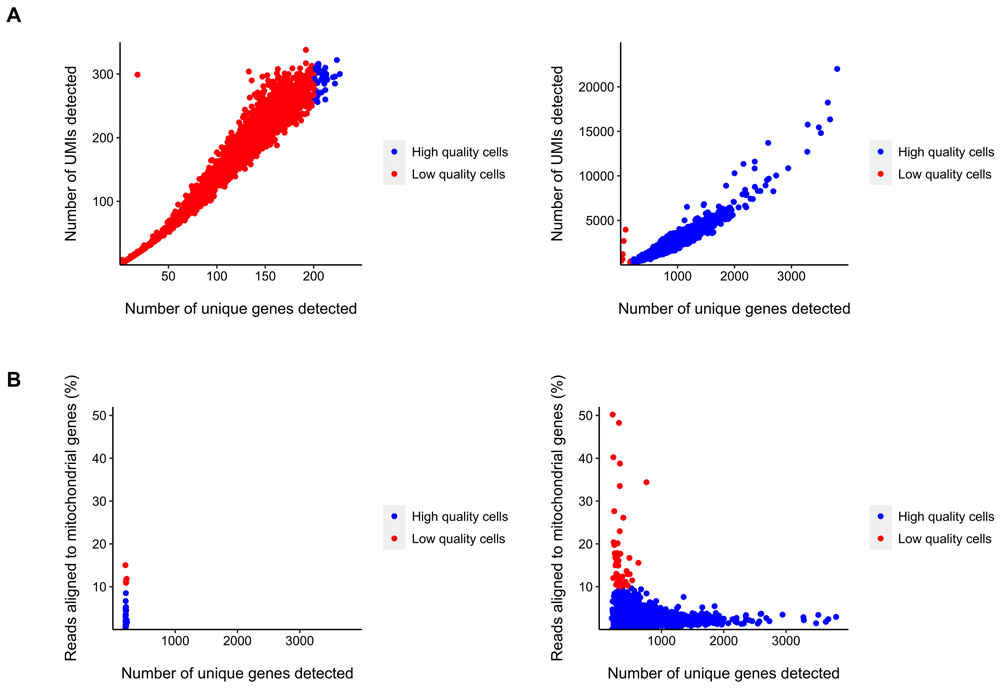
Cells that fall (A) below the given threshold for gene count (in this case, 200) and (B) above the maximum threshold for mitochondrial percentage (in this case, 10%) are shown in red.
Potential cell doublets (black dots) are identified relative to the median total gene count per cell. Cells with a total gene count more than six MADs from the median are flagged for removal.

The total number of UMIs relative to the total number of unique genes detected is visualized before (blue) and after (pink) normalization and variance stabilization using regularized negative binomial regression, as implemented in sctransform (Hafemeister & Satija, 2019). Each point represents a single cell.

The top 2000 most highly variable genes (purple) are identified based on standardized variance. Each dot represents a gene with its standardized variance shown on the y-axis and the average gene expression (calculated using the normalized gene counts) on the x-axis. The 10 genes with the greatest variance across all cells are labelled.
Finally, to demonstrate the quality of the preprocessed data obtained from SASCRiP, Seurat v3.2.0 was used to cluster the two datasets identified as being of high quality (donors B and C) following processing with SASCRiP. The RunPCA function was used to perform dimension reduction (Stuart et al., 2019), and graph-based clustering was performed using the FindNeighbours function (Xu & Su, 2015). In both datasets, the preprocessed data obtained from SASCRiP produced distinct clusters (Figure 6A), corresponding to major PBMC cell types (Figure 6B), suggesting that SASCRiP produces high quality, preprocessed data, suitable for downstream scRNA-seq analyses and applications.

Uniform manifold approximation and projection of ~8000 cells derived from a single donor is shown, coloured according to (A) the clusters identified in the dataset, and (B) the expression of known cell type markers in each cluster. CD3D indicates T cell clusters, MS4A1 B cell clusters, CD68 monocyte clusters, and FCER1A dendritic cell clusters.
SASCRiP is a Python package that provides a simple, streamlined workflow for preprocessing UMI count-based scRNA-seq data through a series of parameterized functions. To ensure flexibility, these functions allow users to either adopt a set of clearly defined default parameters or to modify any or all of these parameters as they see fit. Also, due to its modular design, SASCRiP’s four major functions (kallisto_bustools_count, seurat_matrix, cell_cqc, sctransform_normalize) can be executed either independently to produce custom visualizations and/or intermediary files that can be used as input for other scRNA-seq tools, or all-in-one to produce high-quality, normalized gene expression datasets that are ready to use for downstream analyses. Collectively, these functions seamlessly integrate the functionality of the widely used R packages, Seurat and sctransform, into a custom Python workflow built around kb-python’s implementation of the kallisto | bustools workflow. In this way, pseudoalignment, quantification of gene expression, removal of low quality cells and cell doublets, and normalization and variance stabilization can be performed within a single scRNA-seq analysis pipeline.
All data underlying the results are freely available through 10X Genomics:
Donor A:
https://cf.10xgenomics.com/samples/cell-exp/1.1.0/frozen_pbmc_donor_a/frozen_pbmc_donor_a_fastqs.tar
Donor B:
https://cf.10xgenomics.com/samples/cell-exp/1.1.0/frozen_pbmc_donor_b/frozen_pbmc_donor_b_fastqs.tar
Donor C:
https://cf.10xgenomics.com/samples/cell-exp/1.1.0/frozen_pbmc_donor_c/frozen_pbmc_donor_c_fastqs.tar
Zenodo: SASCRiP Supporting Data https://doi.org/10.5281/zenodo.5899870 (Moonsamy, 2021b)
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
Software available from: https://pypi.org/project/SASCRiP
Source code available from: https://github.com/Darisia/SASCRiP
Archived source code at time of publication: https://doi.org/10.5281/zenodo.5554770 (Moonsamy, 2021a)
License: GNU GPLv3
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)