Keywords
immunosenescence, systemic lupus erythematosus, disease activity, senescence markers
This article is included in the Cell & Molecular Biology gateway.
immunosenescence, systemic lupus erythematosus, disease activity, senescence markers
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with unclear mechanisms that primarily affect women of children-bearing age. Although the pathogenesis of SLE is not well understood, several predisposing factors are involved in the development of aberrant immune response in SLE, including genetic predisposition, gender, and the environment.1 Both innate and adaptive immune systems are involved in the pathogenesis of SLE, which leads to the dysregulation of pro-inflammatory cytokines, autoantibody deposition, oxidative stress, and organs damage.2 Recent studies discovered that several autoimmune diseases possess some immune system abnormalities that resemble the typical characteristics of immune dysfunction described in the elderly, called immunosenescence.3,4 A previous systematic review demonstrates that the shortened telomere length occurs in SLE in relatively young patients.5 Chronic autoantigen stimulation, oxidative stress, or increasing pro-inflammatory cytokines contribute to the replicative stress that accelerates immunosenescence in several autoimmune diseases, including SLE.6,7 These findings indicate that patients with autoimmune diseases may have an accelerated immunosenescence, despite being young.
Aging in the immune system is characterized by several T cell phenotypes and functions changes. Upon repeated activation, T cells progressively downregulate CD28 expression leading to the accumulation of CD28null T cells.8 Moreover, the senescent T cells also express some additional markers such as killer cell lectin-like receptor subfamily G (KLRG-1) and CD57.9 CD45 isoforms (CD45RA or CD45RO) are also aging features in the immune system.10 Terminally differentiated T cells have been described to re-express the CD45RA, while CD45RO can be found abundantly on T cell central memory and effector memory that increases during aging.11,12 Several cytokines also can be applied as descent markers of immunosenescence. Terminally differentiated T cells are capable of producing high levels of interferon γ (IFNγ).13 On the other hand, the level of interleukin 2 (IL-2) is progressively declined in elderly people.14 Chronic infection, such as CMV, has also been associated with immunosenescence in several populations. A systematic review showed that CMV seemed to enhance immunosenescence by inducing highly differentiated effector memory and T cell effector memory re-expressing CD45RA (TEMRA) cells in CD4 and CD8 pools.15
The association between the senescence markers and disease activity in SLE has been described previously but still not given any convincing results yet. Earlier studies indicate that an increasing number of T cell senescence CD4+CD28null correlates with SLICC/ACR damage index in SLE patients.16 Research by Ugarte-Gil et al. found that CD4+CD28null T cells predict the occurrence of new lung damage in SLE.17 Alteration of effector memory T cells (CD45RO+ T cells) was also associated with tissue injury in lupus nephritis.18 Terminally differentiated effector memory T cells (CCR7-CD45RA+) expand in SLE patients with higher disease activity. In addition, SLE patients with higher Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) scores also had a higher percentage of circulating CD4+ T cells with CD28null phenotype.19 Although these senescence markers can be used as potential targets to predict the SLE disease activity, not all senescence markers have been described in SLE patients. Based on these findings, inspecting several senescence markers instead of only one marker at the same time also may increase the probability of predicting SLE disease activity.
The study aimed to identify which senescence markers could be associated with the SLE disease activity. Once the senescence markers were recognized, we would produce a model that showed the best performance to predict the active disease in SLE. We analyzed several senescence markers on both CD4+ and CD8+ T cells compartments, including the senescence surface markers, such as CD28, CD57, KLRG1, and CD45 isoforms (RA/RO); cytokines that associated with aging (IFNγ and IL-2); and IgG anti-CMV antibody. We also compared the performance of these senescence markers with complements and anti-dsDNA as the conventional biomarkers to predict the active disease in SLE.
This research was a cross-sectional study held from January to November 2021. According to the design of this study, the sample size calculation was done according to the formula for cross-sectional study with quantitative variable with the following equation, sample size = (Z1-α/2)2 (SD)2/ (d)2.20 Z1-α/2 value was the standardized value for the confidence interval (CI). Because this study used 95% CI, the Z-score value was 1.96. The value of standard deviation (SD) and d score (margin of error) was obtained and calculated from our previous study that mentioned the SD of CD28null T cells that was 13 and the calculation of d score was 5.4 [Kalim, et al., 2019]. Thus, the minimal sample size of this study was 21 subjects.
In order to minimize the selection bias, the study participants were recruited by simple random sampling. In the end, we recruited 61 female SLE patients referred to the Rheumatology Clinic, Department of Internal Medicine, Saiful Anwar General Hospital, Malang, Indonesia. Patients were approached to take part in this study by several methods, including letter, phone, or direct oral approach in the clinics. Patients who agreed to participate in the study signed the written informed consent. All patients aged 18 to 45 years met the 2019 EULAR/ACR criteria for SLE.21 The exclusion criteria for the subjects were SLE patients who were pregnant, had an active infection, or had a history of cancer.
All subjects were collected for the demographic data, clinical manifestations, laboratory tests, and past medical history by the rheumatologist as the routine procedures in the clinics. To minimize the information bias, patients were assessed by at least two rheumatologists in the clinic. Patients received standard treatment protocols according to the disease severity. As the comparator, 60 healthy individuals were recruited with matched age, sex, and demographic data that were approached by letters or phone. All healthy subjects were stated as “healthy” by at least two doctors, to minimize the bias, from the General Checkup Clinics of Saiful Anwar General Hospital, Malang, Indonesia after underwent clinical and laboratory examinations as a special appointment for this study and did not have any medical problems by history taking.
Subjects signed an informed consent form before undergoing the study, and Saiful Anwar General Hospital Ethical Committee approved all protocols (Ethical Number 400/085/K.3/302/2020 issued on 23rd March 2020).
SLE disease activity was assessed using the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI)-2K score.22 The rheumatologist evaluated the SLEDAI score in the Rheumatology Clinics on each visit. The clinically active disease was defined if the SLEDAI score was >4. In contrast, the clinically inactive disease or lupus low disease activity (LLDAS) was characterized if the SLEDAI score was ≤4 with no activity in major organ systems (renal, central nervous system (CNS), cardiopulmonary, vasculitis, fever) and no hemolytic anemia or gastrointestinal activity.23 The sera of SLE patients were collected during visits and used to measure the C3, C4, and anti-dsDNA examinations. All the laboratory examinations were done in the Central Laboratory Saiful Anwar General Hospital, Malang, Indonesia.
Several cytokines were measured from the sera of SLE patients and healthy subjects, including IFNγ and IL-2. The sera's cytokine levels were measured using the ELISA kits from Biolegend (LEGEND MAXTM human IFNγ ELISA kit [Biolegend, Singapore, cat number 430107] and LEGEND MAXTM human IL-2 ELISA kit [Biolegend, Singapore, cat number 430107]). Serum IgG CMV levels from both subjects were also measured using a human anti-CMV IgG ELISA kit (Abcam, Singapore, cat number ab108724) to examine the role of CMV infection in the development of immunosenescence in SLE patients. All the procedures were done according to the manufacturer’s protocols. In brief, this assay employed the quantitative sandwich enzyme immunoassay technique. A polyclonal antibody specific for IFNγ, IL-2, or CMV antigens has been precoated onto a microplate. Standards and samples are pipetted into the wells and the measured antigens are bound by the immobilized antibody. After washing away any unbound substances, an enzyme-linked polyclonal antibody is added to the wells. Following a wash to remove any unbound antibody-enzyme reagent, a substrate solution is added to the wells and color develops in proportion to the amount of antigen-antibody bound in the initial step. The color development is stopped and the intensity of the color is measured by the microplate reader (Stat Fax 303 Plus microplate reader). The results of IFNγ and IL-2 were measured in pg/ml while IgG anti-CMV was in IU/ml.
Several surface markers associated with the aging of T lymphocytes were measured from the peripheral blood mononuclear cells (PBMC) of SLE patients and healthy subjects. All subjects had 10-15 cc of vein blood taken, then PBMC were isolated from peripheral blood using Lymphoprep (Stemcell Technology, cat number NC0423266) by centrifugation (Hettich model EBA 200) (1600 g for 30 minutes). The formed PBMC layer was taken slowly and rewashed with ten cc phosphate buffer saline (PBS). The supernatant was discarded and centrifuged (Hettich model EBA 200) at room temperature (1200 g for 30 minutes).
The PBMC was stained with the following antibodies: APC/Cy7.7 anti-human CD3, FITC anti-human CD4, PerCP anti-human CD8, PE anti-humanCD28, PE anti-human CD57, PE anti-human CD45RA, PE anti-human CD45RO, and Alexa Fluor 488 anti-Human KLRG1. All kits were purchased from Biolegend, and the procedures were done as the manufacturer protocols. T cells (CD3+ cells) senescence markers which measured were as follows: CD4/CD8 ratio, CD4+CD28null, CD8+CD28null, CD4+CD57+, CD8+CD57+, CD4+CD45RA+, CD8+CD45RA+, CD4+CD45RO+, CD8+CD45RO+, CD4+KLRG1+, and CD8+KLRG1+. The percentages of the cells were acquired with flow cytometry (BD FACScalibur) and analyzed with BD Cell Quest Pro software version 6.0 for Mac. Measurements were made in 10.000 cells, and the results were obtained in the form of percentages (%) of cells.
Statistical analysis was performed using SPSS (IBM SPSS Statistics, RRID:SCR_019096) for Windows version 25.0. Mean ± standard deviation (SD) was used to describe normally distributed data, the median and interquartile range for skewed data, and frequencies of categorical data used the percentages. The Mann-Whitney test compared healthy subjects with SLE patients and active with inactive patients if the data distribution was not normal. Instead, an unpaired t-test was used if the data distribution was normal. Chi-square or Fisher exact test analysis was used to compare two categorical data. Receiver operating characteristics (ROC) curves discriminated active from inactive SLE for each marker. The performance of the senescence markers in predicting the active disease in SLE was shown as the area under curve (AUC). AUC value below 0.5 indicated an inferior model, AUC value more than 0.5 mean that the model is no better at predicting an outcome than random chance, AUC value over 0.7 indicated a good model, values over 0.8 indicated a robust model and a value of 1 means that the model perfectly predicts the outcomes. The optimal cut-off value with the sensitivity, specificity, and likelihood ratio (LR) was also calculated. Logistic regression models were used to identify the independent predictive factors for the active SLE status. The results were considered statistically significant if the p-value was <0.05.
A total of 61 patients and 60 healthy subjects were included in the analysis of this study.41 Study clinicians assessed 128 patients and 83 healthy subjects for eligibility (Figure 1). Healthy subjects were matched for age, race, and sex. Of these, 47 SLE patients (36.7%) and 14 healthy participants (16.8%) did not fulfil the inclusion criteria, 11 patients (8.6%) and four healthy subjects (4.8%) declined to participate. Amongst the enrolled patients and healthy subjects, nine SLE patients (7.0%) and five healthy participants (6.0%) were excluded from the analysis because their records or examination results did not complete and insufficient for being analyzed.
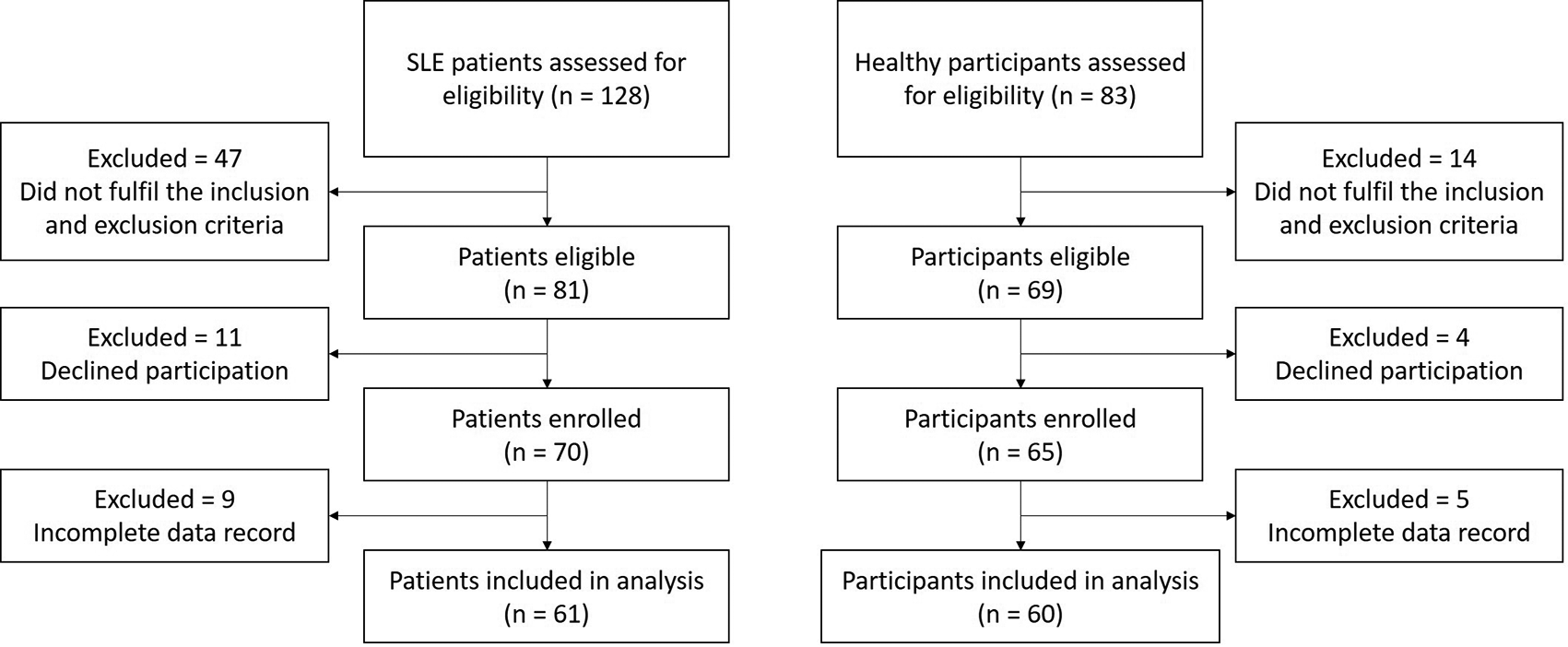
SLE = systemic lupus erythematosus.
According to Table 1, all healthy subjects and SLE patients were women of child-bearing ages, and there was no statistical difference of age between these two groups (p = 0.108). SLE patients were categorized into active and inactive SLE based on the SLEDAI-2K score. Among 61 subjects with SLE, 18 patients (29.5%) were inactive, and 43 patients (70.5%) were active. The median duration of the disease from inactive and active SLE patients was not statistically different (48.0 vs. 44.0 months, respectively, p = 0.384). Age of onset from the first time diagnosed as SLE was also not statistically different between these two groups (24.9 ± 6.1 vs. 23.2 ± 5.6, for inactive and active SLE, respectively, p = 0.282). The mean SLEDAI score from inactive SLE was 1.2 ± 1.0, while active SLE was 11.3 ± 8.1 (p = 0.000). The significant differences in clinical manifestations between inactive and active SLE were renal involvement, serositis, and vasculitis. In total, 20 patients from the active groups showed symptoms of lupus nephritis (p = 0.001), while ten patients from active SLE groups had symptoms of vasculitis and serositis (p = 0.039). All patients from inactive and active SLE groups received standard treatment based on their disease activity. There were no significant differences in the treatment distribution from both groups (Table 1).
Before determining the possible senescence markers that could predict the disease activity in SLE, we analyzed the comparison between these markers between healthy subjects and SLE patients to find which senescence markers significantly changed in SLE populations. According to Table 2, we demonstrated that SLE patients had a significantly lower CD4/CD8 ratio compared to healthy subjects (0.6 ± 0.2 vs. 1.5 ± 0.7, p < 0.001). Among all senescence markers measured in the CD4 T cell population, CD28null, CD45RO, and KLRG1 were significantly higher in SLE patients than healthy subjects (p < 0.001 for all markers). On the other hand, all senescence markers from the CD8 T cell population, including CD28null, CD57, CD45RA, CD45RO, and KLRG1, were significantly higher in SLE patients (p < 0.001 for all markers). Examination of the cytokines level revealed that serum IFNγ level was markedly higher in SLE patients (221.7 ± 137.5 pg/ml vs. 7.1 ± 2.8 pg/ml, p < 0.001). In contrast, a significantly lower IL-2 level was demonstrated in SLE patients compared to healthy subjects (13.9 ± 7.5 pg/ml vs. 228.3 ± 132.5 pg/ml, p < 0.001). We did not find significant differences in IgG CMV levels between SLE patients and healthy subjects (p = 0.512). According to Table 2, markers that did not statistically differ between the healthy subjects and SLE patients were excluded for the subsequent analysis.
In the next analysis, we compared the senescence markers between active and inactive SLE patients according to the SLEDAI-2K score (Table 3). Our findings showed that among CD4 T cell senescence markers, none of them was statistically different between active and inactive SLE patients. In contrast, all of senescence markers from CD8 T cell populations were significantly higher in active patients compared to inactive patients with SLE, including CD28null (16.1 ± 7.4% vs. 8.4 ± 5.9%, p = 0.001), CD57 (11.9 ± 5.8% vs. 6.3 ± 4.6%, p = 0.001), CD45RA (25.0 ± 9.5% vs. 19.6 ± 6.6%, p = 0.043), CD45RO (11.1 ± 7.7% vs. 6.4 ± 4.6%, p = 0.024), and KLRG1 (14.6 ± 7.1% vs. 7.6 ± 4.7, p < 0.001). Markedly lower of CD4/CD8 ratio was found in active SLE patients compared to the inactive patients (0.6 ± 0.2 vs. 0.7 ± 0.2, p = 0.026). Significantly higher IFNγ levels were demonstrated in SLE patients with active disease activity (241.3 ± 153.4 pg/ml vs. 171.1 ± 62.8 pg/ml, p = 0.043).
We also analyzed the anti-dsDNA, C3, and C4 serum levels as control markers, because those markers had been established as standard markers to monitor the disease activity in SLE. The anti-dsDNA level was statistically higher in SLE patients with active disease (138.9 ± 88.4 IU/ml vs. 76.5 ± 72.4 IU/ml, p = 0.043). On the other hand, C3 and C4 levels were statistically lower in active SLE patients than patients with inactive disease activity (p = 0.015 and p = 0.004, respectively). All markers that did not statistically differ from the inactive SLE patients were excluded for the subsequent analysis.
The ROC curve analysis showing the AUC for each marker in predicting the SLE active disease is presented in Table 4. C3, as the control marker, had the highest AUC value compared to other markers (AUC 0.844). As for the senescence marker, CD8+CD28null had the highest AUC compared to other senescence markers (AUC 0.801). The optimal cut-off value with the sensitivity, specificity, and LR is shown in Table 4. CD8+CD28null had the highest sensitivity (91.9%) compared to other markers with the lowest negative likelihood ratio (LR- = 0.13) in the cut-off of >6.85%. The highest specificity was possessed by C3 (75%) in the cut-off <0.6 g/l with the highest positive likelihood ratio (LR+) of 3.09. The senescence markers from the CD8 T cell population, including CD28null, CD57, CD45RA, CD45RO, and KLRG1, had higher sensitivity than anti-dsDNA, C3, or C4.
Univariate and multivariate analysis using the logistic regression for the markers to predict the active disease of SLE is presented in Table 5. We used the cut-value from the prior ROC analysis to define the upper and lower value from the markers. The univariate analysis yielded statistically significant OR for active SLE by the following markers: CD4/CD8 ratio, CD8+CD28null, CD8+CD57+, CD8+CD45RA+, CD8+CD45RO+, CD8+KLRG1+, IFNγ, C3, and C4. According to the multivariate analysis, CD4/CD8 ratio (OR 9.1 [95% CI 2.2 – 37.6], p = 0.002), CD8+CD28null (OR 25.5 [95% CI 5.1 – 128.2], p < 0.001), and C3 (OR 17.9 [95% CI 2.1 – 155.2], p = 0.006) had significant association with the active disease of SLE from the participants.
According to the multivariate analysis, we demonstrated that CD4/CD8 ratio, CD8+CD28null, and C3 yielded a significant association with active disease of SLE in our subjects. Thus, our subsequent analysis was to observe the performance of the combined model of these markers to predict the active disease of SLE. The ROC curve analysis of the combined marker showing the AUC, sensitivity, specificity, and LR was presented in Table 6. Overall, using the combined marker models had a better AUC value compared to the previous single marker in predicting the active disease of SLE. The combination of T cell senescence markers, CD4/CD8 ratio, and CD8+CD28null had an AUC value of 0.859 with a sensitivity of 87.5% and specificity of 64.3%. The best AUC value was demonstrated from the combination model of CD4/CD8 ratio, CD8+CD28null, and C3 (AUC 0.923) with sensitivity of 81.2% and specificity of 84.0%. Our finding also showed that the sensitivity, specificity, and LR+ from the combination model from all three parameters were higher than standard C3, C4, or anti-dsDNA as control markers for disease activity in SLE.
Immunosenescence is a dynamic alteration of immune systems related to the aging process.24 Although immunosenescence has been studied in all immune system cells, CD4 or CD8 T cell senescence is the primary immune aging component associated with several pathologies.25 Several markers that are associated with the T cell senescence have already been reported, including the surface markers (CD28null, CD57, CD45 isoforms, or KLRG1) and cytokines (IFNγ or IL-2).8–15 Although several immunosenescence markers have been discovered, not all of these markers are exclusively studied in SLE. Our findings demonstrated significant changes in the T cell senescence markers in the SLE patients compared to healthy individuals of similar age. We showed that all senescence markers from CD8 T cell surface (CD28null, CD57, CD45 isoforms, and KLRG1) were significantly higher in the SLE patients; instead, only some surface markers were elevated in the CD4 T cell populations (CD28null, CD45RO, and KLRG1). These findings suggested that CD8 T cells might be more susceptible to aging compared to the CD4 T cells in SLE.
Some evidence suggested that CD4 and CD8 T cells behave differently in response to aging. A previous study showed that CD4 T cells might be a more stable cell type, with lesser susceptibility to age-dependent phenotypic and functional change.26 In contrast, CD8 T cells were intrinsically more susceptible to phenotypic changes related to aging. For instance, the rate of CD28 loss in CD8 T cells was faster than in the CD4 T cells populations. A phenotypic shift of the central memory T cells was more pronounced in CD8 T cells than CD4 compartments.27 In addition, senescent CD8 T cells reduced their apoptosis capability and diminished caspase 3 activity.28 This phenomenon would increase CD8 T cells, whereas CD4 T cells were decreasing in aging populations, characterized by the inverse ratio of CD4/CD8 T cells. Consistent with the previous study,29 our results also showed an inverse ratio of CD4/CD8 T cells in SLE patients.
The relationship between the disease activity and the aged CD8 T cells in SLE might be because of the ability to secrete the pro-inflammatory cytokines, such as IFNγ, which could worsen the inflammatory responses. The role of IFNγ for disease activity and organ damages in SLE had been reviewed previously.30 Our analysis found that IFNγ levels were significantly higher in SLE patients with active disease. Similar to our study, memory T cells (central and effector memory) were capable of producing high levels of IFNγ instead of other cytokines (TNFα, IL-4, or IL-5).13 Expanded CD8+CD57 subpopulation also correlated with the increase of IFNγ levels in another study.31
Most studies emphasized the importance of chronic viral infection, such as CMV infection, in the progression of immunosenescence during aging. The inflammatory response initiated by CMV can lead to the immune system remodeling contributed to the aging process.32 However, we showed that IgG CMV antibodies were similar in both SLE patients and healthy subjects. These results might explain that there was no role of CMV infection in developing immunosenescence in SLE.
Although some senescence markers were significantly higher in active SLE patients, head-to-head comparison between the senescence markers with conventional markers (complement or anti-dsDNA) to predict the disease activity in SLE was never described in the previous study. Our data demonstrated that CD8+CD28null showed a higher sensitivity with comparable AUC to predict the active SLE than complement or anti-dsDNA levels. Among the senescence markers analyzed in this study, only CD4/CD8 ratio and CD8+CD28null had significantly increased OR from the multivariate analysis. In addition, C3 levels also had an increased OR significantly from the multivariate analysis. Our study revealed that using a combination of the senescence markers (CD4/CD8 ratio and CD8+CD28null) with the C3 levels to predict the active disease in SLE produced the highest result AUC with better sensitivity, specificity, and LR value compared to single-use of the marker.
The CD4/CD8 ratio and CD8+CD28null were already described in the previous study as immune risk profile (IRP). IRP consisted of several laboratory markers: a low CD4/CD8 T-cell ratio, an expansion of CD8+CD28- T-cells, and cytomegalovirus (CMV) seropositivity.33 In contrast to our present study, we did not find any association between CMV seropositivity with the disease activity in SLE. The presence of IRP characterized by the low CD4/CD8 ratio and expansion of CD8+CD28null T cell in the elderly was associated with poor life expectancy and increased mortality or morbidity.34,35 Unlike our study, Ugarte et al. demonstrated that, instead of CD8+CD28null T cells, CD4+CD28null T cells predicted lung damage in SLE patients with hazard ratio (HR) 1.042.17 Another study showed that terminally differentiated CD8 T cells were associated with renal pathology in patients with lupus nephritis.36 In addition, Winchester et al. also showed that CD8 CD28null T cells were mainly found on the kidney tissue of lupus nephritis patients, indicating the possible role in promoting tissue injury in lupus nephritis.37 Despite that, the role of aged CD8 T cells in non-renal SLE manifestation was never described.
In our understanding, this is the first study that described the role of several immunosenescence markers in predicting the active disease of SLE. We also demonstrated that by combining the senescence and conventional markers could provide a better value in predicting the active disease of SLE. In addition, SLE is a complex autoimmune disease with multiple pathways of pathogenesis. Therefore, by understanding some other mechanisms that led to the disease progressivity in SLE, we hope that this disease can be more preventable and curable in the future. However, there are still limitations in this current study. We still could not explain the possible mechanism on how the T cell senescence might affect the disease severity of SLE. A previous study demonstrated that the senescent T cells might affect the disease activity by secreting the inflammatory cytokines.38 However, we did not demonstrate any association of IFNγ with the disease activity in this study. IFNγ is an abundance cytokine produced by several cells, not only the senescent T cells. Therefore, it is not clear that the increase of the IFNγ in this study was solely caused by the senescent T cells. However, previous studies still showed the role of pathogenic senescent T cells in promoting inflammation in certain conditions.39,40 Thus, we still suspected that there might be other cytokines or molecules associated with the disease progression in SLE because of the T cell senescence.
In conclusion, all analyses demonstrated that two senescence markers (CD4/CD8 ratio and CD8+CD28null) predicted the SLE disease activity with good sensitivity and specificity. Combined with the conventional markers of C3 might better predict the active disease of SLE. However, the progressivity of disease activity in SLE is a dynamic process; therefore, a longitudinal study over a long period is still needed to convince the role of immunosenescence in the disease progression of SLE. These findings may help to understand better the immunopathogenesis of SLE, and also, these senescence markers may be potential targets as a diagnostic or prognostic marker or even as a marker to monitor the therapy.
Figshare: Data. https://doi.org/10.6084/m9.figshare.19100306.41
Data are available under the terms of the Creative Commons Attribution 4.0 International license (CC-BY 4.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)