Keywords
Transcriptional regulation, rSNP, TF-DNA binding, SNP-SELEX, PWM, PSSM
This article is included in the Bioinformatics gateway.
Transcriptional regulation, rSNP, TF-DNA binding, SNP-SELEX, PWM, PSSM
The reviewer has drawn our attention to the missing citation of one of the key PWM benchmarking papers. We also updated the link to supplementary materials.
See the authors' detailed response to the review by Victor G. Levitsky
See the authors' detailed response to the review by Philip Machanick
Gene regulatory regions constitute an important part of non-coding DNA which defines both the global development program of a mammal and individual traits of a particular organism. Specific recognition of DNA sites by transcription factors (TFs) provides the gear system linking individual genomic variants to phenotypes.1 The commonly accepted model to quantify the specificity of transcription factor binding to various DNA sites is the position weight matrix (PWM), which specifies additive contributions of individual nucleotides to the protein-DNA binding energy.2,3 Recently Yan et al.4 presented a powerful high-throughput experimental technique, SNP-SELEX, which allows measuring differential TF binding to alternative alleles in vitro. Yan et al. used the experimental data they had obtained for many TFs to assess the performance of PWM in predicting differential TF binding to alternative alleles and compare it to that of deltaSVM, a more complex method based on machine learning. As a result, they reported that in this setting “the position weight matrices of most transcription factors lack sufficient predictive power”. Keeping in mind that PWMs are extensively used for prediction of the regulatory potential of single-nucleotide variants5–8 the finding of Yan et al. could be devastating for a vast array of research projects and software tools.
Yan et al. tend to explain the poor performance of PWMs by model limitations, primarily, arising from the oversimplistic assumption that nucleotides occupying different positions in the binding site provide independent contributions to the binding energy. Here we re-analyze the dataset of Yan et al. and argue that the poor PWM performance in predicting differential transcription factor binding to alternative alleles detected by SNP-SELEX is to a major extent explained not by the principle limitations of PWM as a mathematical construction but rather by particular inadequate PWMs for TFs under study. We show that the careful selection of PWMs of many TFs from a public database quantitatively explains the differential TF binding to allelic variants with reliability comparable to deltaSVM.
To re-assess PWM performance, we used PWMs stored in the CIS-BP database,9,10 which contains PWMs constructed from data obtained with different experimental techniques for thousands of TFs for different species. With the objective of selecting the PWM appropriate for quantifying differential allele binding of a TF, for each of 129 TFs assessed in Yan et al. we extracted an extended set of candidate PWMs, with a median of 32 PWMs per TF. The overall distribution was non-uniform e.g. there were only 2 candidate PWMs for ZNF396 and over a thousand for FOXA2, see Extended data, Supplementary Table S1.
Through cross-validation on the 1st batch of SNP-SELEX data following the strategy of Yan et al., we selected the best PWMCIS-BP for each TF (see “Selecting the best PWMs and estimating PWM performance with SNP-SELEX data” in the Methods). There was no correlation between the prediction performance (area under precision-recall curve, AUPRC) and the number of tested PWMs per TF (r = −0.07, P = 0.425). Many of the best-performing PWMs were originally constructed from the data related not to the target TF but to other TFs sharing the same DNA-binding domain as the TF of interest. Some PWMs were based upon the TF binding data from different species. We denote by ΔPWMCIS-BP the difference of the allelic scores predicted with PWMCIS-BP and by ΔPWMMult the results of PWMs obtained from HT-SELEX data using the multinomial algorithm11 and assessed by Yan et al.4
Yan et al. provide the experimental differential binding scores for each SNP (the “pbSNP” scores). We compared these scores with ΔPWMCIS-BP predictions. For a complete set of 816594 TF-SNP pairs (Figure 1a), the Pearson correlation coefficient was comparable to that observed by Yan et al. (0.531 vs 0.534 in Yan et al., see their Figure 2a). Yet, if only SNPs with a strong predicted binding are included (P < 10-4 for the PWM score of a stronger bound allele) then a much higher correlation (r ~ 0.828) is achieved, see Figure 1b. Finally, if only the most strongly bound TF is considered for each SNP (as in Figure 3a of Yan et al.), the respective correlation reaches 0.711, comparable to -0.777 reported for deltaSVM in Yan et al. (compare to their Figure 1c).
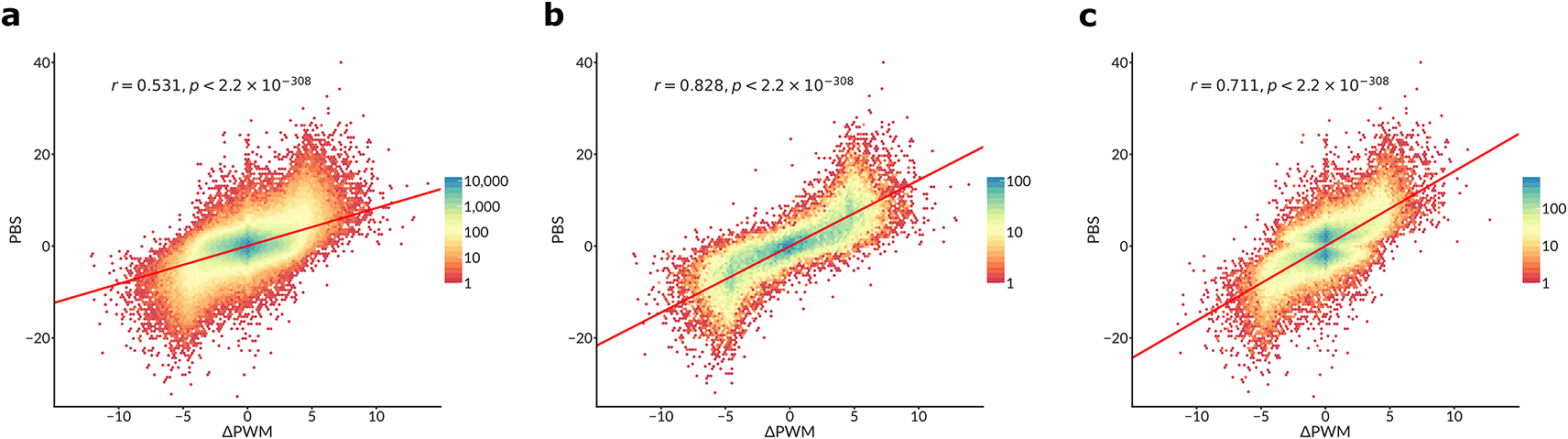
Hex-plots of PBS scores (Y-axis) vs ΔPWMCIS-BP predictions (X-axis) for different sets of SNPs, analogous to Figure 2a and Figure 3a in Yan et al., with PCC and two-sided t-test P-values displayed.
a. Complete set of 816594 TF-SNP pairs for 129 TFs that were used to compare the performance of deltaSVM vs ΔPWM in Yan et al.
b. A subset of 35837 TF-SNP pairs overlapping strong PWM hits (P < 10-4 for a stronger bound allele).
c. A subset of 84962 TF-SNP pairs obtained by considering only the TF with the highest PBS score for each SNP (as in Figure 3a of Yan et al).
To assess ΔPWMCIS-BP performance at varying binding affinity ranges we, similarly to Yan et al., categorized the SNPs into five quantiles based on their observed affinities (“OBS” scores) and assessed the performance of ΔPWMCIS-BP separately for each quantile. For all quantiles but the lowest (the weakest bound sites) ΔPWMCIS-BP outperformed ΔPWMMult of Yan et al. Notably, the performance of ΔPWMCIS-BP was especially high for the middle quantiles and for the highest quantile was on par with deltaSVM (see Supplementary Figure S1, compare with Extended data Figure 7 in Yan et al.). Particularly, for strongly bound SNPs from high quantiles in the first SNP-SELEX batch, ΔPWMCIS-BP did not display any TFs with a very small AUPRC (i.e. prediction failures), the other metric for which deltaSVM dramatically outperformed ΔPWMMult.
Next, we compared the overall performance of ΔPWMCIS-BP for different TFs at the 1st SNP-SELEX batch. For more than a half (72 of 129) of transcription factors ΔPWMCIS-BP achieved reliable predictions fulfilling the same criterion as in Yan et al. of the AUPRC > 0.75, see Figure 2a. This is 3 times more transcription factors with reliable PWM predictions than reported in Yan et al. for ΔPWMMult (only 24 out of 129). Notably, we obtained good predictions in some cases reported as markedly underperforming such as FOXA2 (compare Figure 2b with Figure 2b of Yan et al.). Another TF performing markedly poorly for PWMMult was IRF3, but the best PWMCIS-BP performed better than both ΔPWMMult and deltaSVM (AUPRC for ΔPWMCIS-BP of 0.298 as compared to 0.184 of deltaSVM). In some cases, the predictive power of ΔPWMCIS-BP went in line with that of ΔPWMMult, for instance, TFAP transcription factors in both cases displayed outstanding performance (AUPRC of 0.9 for ΔPWMMult and 0.92 for ΔPWMCIS-BP) whereas E2F family transcription factors in both cases performed worse (AUPRC of 0.4 for ΔPWMMult and 0.42 for ΔPWMCIS-BP).
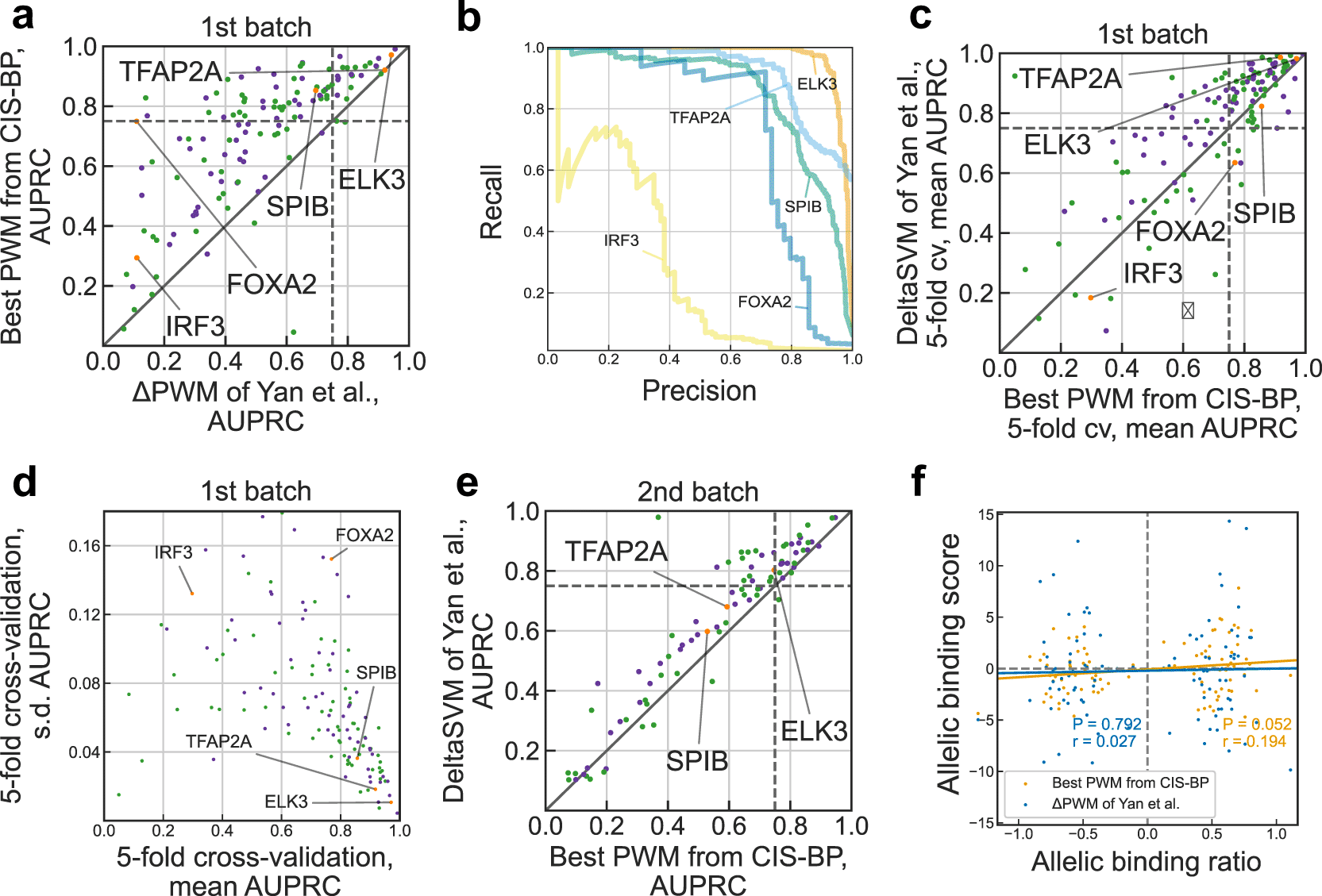
a. Comparison of performance of Yan et al. ΔPWM (x-axis) and best CIS-BP position weight matrices (PWMs) in predicting preferential binding SNPs in the 1st batch on the SNP-SELEX data. Each point denotes one of 129 TFs, violet and green points denote inferred and direct PWMs, respectively (see the Methods). Both axes show area under the precision-recall curve (AUPRC) values. Transcription factors (TFs) shown in Figure 2b of Yan et al. are highlighted in orange and labeled. Dashed lines denote AUPRC of 0.75.
b. Examples of the precision-recall curves showing performance of different PWM models in predicting preferential binding SNPs (single-nucleotide polymorphisms) as in Figure 2b of Yan et al.
c. Comparison of performance of deltaSVM (y-axis) and best CIS-BP PWMs (x-axis) in predicting preferential binding SNPs identified in the 1st batch of SNP-SELEX. Each point denotes one of 129 TFs, violet and green points denote inferred and direct PWMs, respectively. Both axes show mean AUPRC values obtained by 5-fold cross-validation (cv). Dashed lines denote AUPRC of 0.75.
d. Variance of performance of CIS-BP PWMs (x-axis: mean AUPRC, y-axis: s.d.) in 5-fold cross-validation using the complete data of the 1st batch of SNP-SELEX. Each point denotes one of 129 TFs, violet and green points denote inferred and direct PWMs, respectively.
e. Comparison of performance of deltaSVM (y-axis) and best CIS-BP PWMs (x-axis) in predicting preferential binding SNPs identified in the 2nd batch of SNP-SELEX. Each point denotes one of 87 TFs, violet and green points denote inferred and direct PWMs, respectively. Both axes show AUPRC values. Dashed lines denote AUPRC of 0.75.
f. Correlation of allelic biases of DNA binding detected from ChIP-Seq experiments in HepG2 cells by Yan et al. and those predicted by ΔPWM of Yan et al. (blue) and best CIS-BP PWMs (orange). Pearson correlation coefficient (r) and the respective P-value are shown. The allelic binding ratio is computed as in Yan et al.; 101 transcription factor-SNP pairs involving 68 unique SNPs and 6 transcription factors (ATF2, FOXA2, HLF, MAFG, YBX1, and FOXA1) are shown.
In fact, for 34 transcription factors, PWMCIS-BP outperformed advanced models of deltaSVM (Figure 2c). 5-fold cross-validation showed that models reaching higher AUPRC simultaneously had a lower variance in prediction quality across individual folds (Figure 2d). Furthermore, we tested the PWMs on the independent 2nd batch data (Figure 2e, compare with Figure 3d of Yan et al.), and it also showed competitive albeit lower performance, with 36 of 124 transcription factors passing 0.75 AUPRC. Finally, we tested if the PWM predictions agreed with the allelic binding ratios in HepG2 ChIP-seq data and found a small but marginally significant correlation (Figure 2f, r = 0.194, P = 0.052) for 101 SNPs tested in Yan et al. and reaching r = 0.235 (P = 0.047) for a subset of 72 SNPs with significant PWMCIS-BP hits (motif P-value < 0.005), in contrast to almost zero correlation for ΔPWMMult reported in Yan et al.
Our approach mimics a machine learning setup, where the best model is selected ("trained") through cross-validation on a first experimental data set (1st batch of the SNP-SELEX data), and then additionally independently validated on the second experimental data set (2nd batch of SNP-SELEX data). As we select from a finite and typically small set of candidate PWMs, the risks of overfitting are minimized, and the resulting performance was not correlated with the number of ‘candidate’ PWMs. The utilized layout allowed us to pick up the best suited PWM independently from the original data or motif discovery method used for PWM construction, yet maintaining the main PWM limitations, such as the assumption of the independent contributions of nucleotides at different TFBS positions.
The lower performance of PWMs for TFBS recognition as compared with more complex models was reported in many publications.12–14 The popular explanation blames the assumption of positional independence of PWM scores, which comes short of taking into account the marked correlations of nucleotides located at neighboring or even distant positions of binding sites.15–17 This shortcoming is also considered over-restrictive for PWM applications in predicting the effects of single nucleotide variants on TF binding,4,18 which has recently come into the spotlight of modern genetics where the advent of complete genome sequencing brought about the need for interpretation of phenotypes associated with regulatory variants.19,20 Here we suggest an alternative explanation of inadequate PWM performance in predicting the effects of single-nucleotide variants on TF binding. In many cases, the reason is an inadequate PWM construction or selection procedure.
Besides technical difficulties in proper “training” a PWM through motif discovery from different types of experimental data, the particular experimental context may influence the applicability of the resulting PWM. Careful selection of PWMs from a pool of alternative existing models results in an apparent improvement of the quantitative assessment of preferential binding to single-nucleotide variants, especially for high-scoring TFBS. In many cases, the prediction power becomes comparable to that of the significantly more complex model such as deltaSVM. We specifically emphasize that in our study all selected PWMCIS-BP were genuine PWMs following the classic assumption of the independent contribution of positional scores.
Summing up, our results do not compromise the high performance of deltaSVM,12 used by Yan et al. as an advanced substitution of position weight matrices (PWMs). However, properly selected PWMs achieve performance that is very close and in some cases even better than that of deltaSVM. Despite the simplicity of the PWM model, its construction is not trivial and its success depends both on the motif discovery algorithm and reliability of the training data. In our case, almost half of the best PWMs were derived from related TFs, including 8 cases of PWMs based on experimental data from other species. The experiments used to obtain the best PWMs were also of different types, including ChIP-Seq, protein-binding microarrays, and SMiLE-Seq data, see Extended data, Supplementary Table S1.21 Thus, it is important to consider various sources of PWMs and select those the most suitable by proper benchmarking. In the context of applying PWMs to analyze regulatory variants, SNP-SELEX of Yan et al. provides rich, unique, and practically useful data.
The objective of our study is by no means to undermine the necessity of complex TFBS models with dependent positional contributions. Advanced multiparametric and alignment-free approaches such as deltaSVM appear very likely to shape the oncoming future of transcription factor binding site models. Rather, we want to underline that the prediction performance of transcription factor binding sites in its current stage is more influenced by model training protocols than by model structure restrictions. PWMs still can deliver a solid standard in representation and bioinformatics analysis of the transcription factor binding sites, including assessment of the functional impact of single nucleotide variants in gene regulatory regions. In addition, we underline that better defined ‘baseline’ PWMs or PWM selection procedures are required for the proper evaluation of advanced models. It is important that such ‘baseline’ TFBS models, while certainly being handicapped by design, still reach meaningful prediction quality. These are good news for thousands of researchers who still use the ‘legacy’ PWM scoring for practical applications in regulatory genomics and bioinformatics.
As a source of candidate PWMs we used the CIS-BP (Catalog of Inferred Sequence Binding Preferences) collection9,10 of pre-made matrices. For each TF, we gathered all PWMs assigned to the TF and added PWMs for related proteins sharing similar DNA binding domains. This was motivated by the results of the benchmarking study of Ambrosini et al.2 where a PWM for some TF often displayed poorer TFBS recognition power than a PWM for some different TF but with the same DNA-binding domains.
The starting set of position frequency matrices was extracted from TF_Information_all_motifs.txt of CIS-BP 2.0 that includes models derived from direct experimental data for each TF and models that can be inferred given the TF family-specific threshold on DNA-binding domain similarity, see Ref. 11. In Figure 2 such PWMs are referred to as ‘direct’ and ‘inferred’. All position frequency matrices were converted to log-odds PWMs as in Ref. 22 with an arbitrarily selected word count of 100, a pseudocount of 1, and uniform background nucleotide probabilities. For each TF, the set of PWMs was additionally extended by considering related TFs, i.e. PWMs for all ETV* TFs were added to the ETV1 PWM set, all FOX* (Forkhead box) PWMs were added to the FOXA2 PWM set, etc. (e.g. YY1 and YY2 PWM sets were identical). This procedure was not performed for ZNF* (zinc finger) TFs as these TFs can recognize very dissimilar motifs and thus additional PWMs of other ZNFs would unlikely provide any benefit.
To assess with a particular PWM whether an SNV affects transcription factor binding, we used PERFECTOS-APE 6 that estimates the log-fold change of motif P-values computed for best PWM hits detected among sites overlapping the first and the second of two alternative alleles. To use the prediction as a binary classifier, we treated the cases with P > 0.005 at both alleles as predicted negatives and used the log-fold change as the prediction score in the remaining cases. The auc function of the sklearn.metrics Python package was used to estimate the area under the precision-recall curve (AUPRC).
To provide a fair assessment, we mimicked the benchmarking protocol of Yan et al. Particularly, true positives and true negatives were selected from the SNP-SELEX data as follows. 1st batch data positives: PBS P-value < 0.01 and OBS P-value < 0.05; negatives: PBS P-value > 0.5 and OBS P-value < 0.05. 2nd batch data positives: PBS P-value < 0.01, negatives: PBS P-value > 0.5. For each TF, we tested each CIS-BP PWM from its PWM set. For each TF, the PWM reaching the highest AUPRC on the 1st batch data was selected for evaluation against the best PWM on the 1st batch (Figure 2a) and against deltaSVM on the 2nd batch of SNP-SELEX data (Figure 2e). Performance estimates for deltaSVM models (used in Figure 2c,e) were extracted from Supplementary Table S7 of Yan et al. Performance estimates of ΔPWMMult (used in Figure 2a) were kindly shared on our request by the authors.4 We also mimicked the stratified five-fold cross-validation procedure used by Yan et al. The mean of AUPRC across the folds was used to compare the performance of ΔPWMCIS-BP with deltaSVM of Yan et al. at the first batch of SNP-SELEX data (Figure 2c).
The data on allelic binding ratios at individual SNPs and respective ΔPWM predictions of Yan et al. (Figure 2f, compare to Figure 2d of Yan et al.) were kindly shared on our request by the authors. The data included 193 TF-SNP pairs demonstrating allelic imbalance with 101 of 193 pairs annotated with the ΔPWM predictions. For these SNPs, we obtained PWM predictions with the same protocol as for the SNP-SELEX data using the best PWMs selected with the 1st batch of the SNP-SELEX data.
Original data on preferential binding SNPs as well as ΔPWM and deltaSVM predictions are provided in the supplementary materials section of the Yan et al. paper.4
CISBP Human PWMs collection was extracted from CIS-BP 2.0.9,10
Figshare: PWM-evaluation-using-SNP-SELEX, https://doi.org/10.6084/m9.figshare.16906789.v1.21
This project contains the following extended data:
• Supplementary table S1 (Overview of PWMs and their performance in recognizing SNPs affecting transcription factor binding in SNP-SELEX data.)
• Supplementary figure S1 (Performance of ΔPWMCIS-BP in predicting weak and strong TF binding sites.)
Data are available under the terms of the Creative Commons Zero “No rights reserved” data waiver (CC0 1.0 Public domain dedication).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)