Keywords
Pestalotiopsis, genome assembly, nanopore sequencing, diy
This article is included in the Bioinformatics gateway.
This article is included in the Genomics and Genetics gateway.
This article is included in the Nanopore Analysis gateway.
Pestalotiopsis, genome assembly, nanopore sequencing, diy
Accessibility to genome sequencing has typically been limited to large, well-funded institutions due to large capital requirements of DNA sequencing platforms, like many high-throughput Illumina platforms that cost hundreds of thousands of dollars to obtain. However, the recently developed MinION nanopore sequencing platform by Oxford Nanopore Technologies provides both the hardware and reagents needed for genome sequencing for $1000 USD, reducing the financial barrier to DNA sequencing. Other equipment like thermocyclers are also becoming more portable and affordable,1 reducing the barrier for “do-it-yourself” (DIY) biologists to perform genetic analysis and engineering projects.2 The distribution of resources and knowledge between DIY biologists and institutional researchers can advance the rate of development to research and provide lower-cost approaches.3
A significant portion of the approximately 22,000 bioactive microbial metabolites are of fungal origin.4 Furthermore, of the estimated three million species of fungi, only about 100,000 species have been identified.5 This poses an opportunity to discover and characterize new metabolite-producing fungi that may lead to the commercialization of novel pharmaceuticals, pigments, enzymes and food-production platforms. Pestalotiopsis, the focus of this study, is a diverse genus of Ascomycete fungi, often pathogenic to plants6 and commonly isolated as endophytes.7 Pestalotiopsis is known to possess potentially valuable metabolic capabilities, including the production of bioactive secondary metabolites like taxol8 and the ability to biodegrade polyurethane.9
The purpose of this work is 1) to better understand the genome of Pestalotiopsis and 2) to demonstrate that high-quality eukaryotic genome sequencing is now accessible in a DIY environment. To do this, we built a high-quality genome of Pestalotiopsis using nanopore sequencing, highlighting the increased accessibility and affordability of genome sequencing for amateur biologists and early-stage startups.
A wild-type strain of Pestalotiopsis was isolated from a wild-type clone of a Laetiporus sp. found growing on an aged hardwood stump at Whiting Ranch Wilderness Park, Orange County, California, United States.
The isolate was maintained on Petri dishes (eBay) containing Malt Extract Agar (MEA) (Amazon.com) media at room temperature in natural indoor ambient light. Microscopic images are shown in Figure 1.
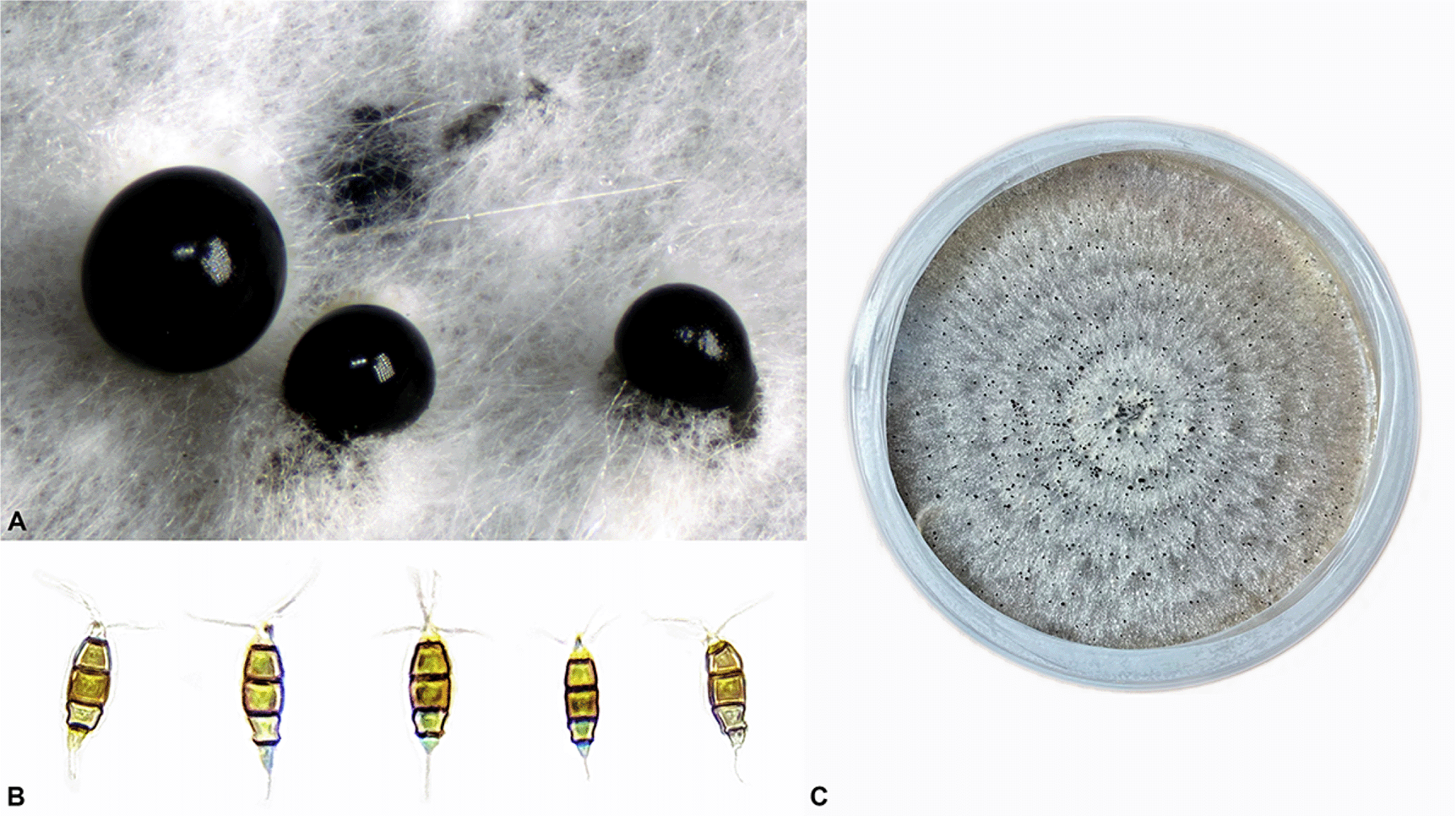
A. Conidiomata B. Conidia C. Mature colony on MEA.
A sterile toothpick was used to aseptically transfer a small swath (2-4 mm) of fresh, non-sporulating Pestalotiopsis mycelium from the surface of solid MEA culture into a 0.2 ml microcentrifuge tube containing 30 l of 0.5 M NaOH (Home Science Tools, SKU: UN1823, MT) lysing buffer.10
A makeshift pestle (Figure 2) was crafted from a 200 l long pipette tip and quickly melted with the flame from a lighter followed by immediate application to a flat surface sanitized with 70% isopropyl alcohol, in order to form a small flat non-porous tip roughly 1 mm in size. Using the makeshift pestle, the tissue was crushed and macerated in the tube for one to three minutes, or until the tissue was homogeneously suspended in the lysis buffer and no clumps were observed.

A. DIY Magnetic Bead Separator Rack constructed with neodymium magnets; B. Vibrating Back Massager as DIY Vortex Mixer; C. Makeshift Pestle generated by melting a pipette tip with over a flame and applying the melted tip to a sterile solid surface before hardening into a flat blunt-end.
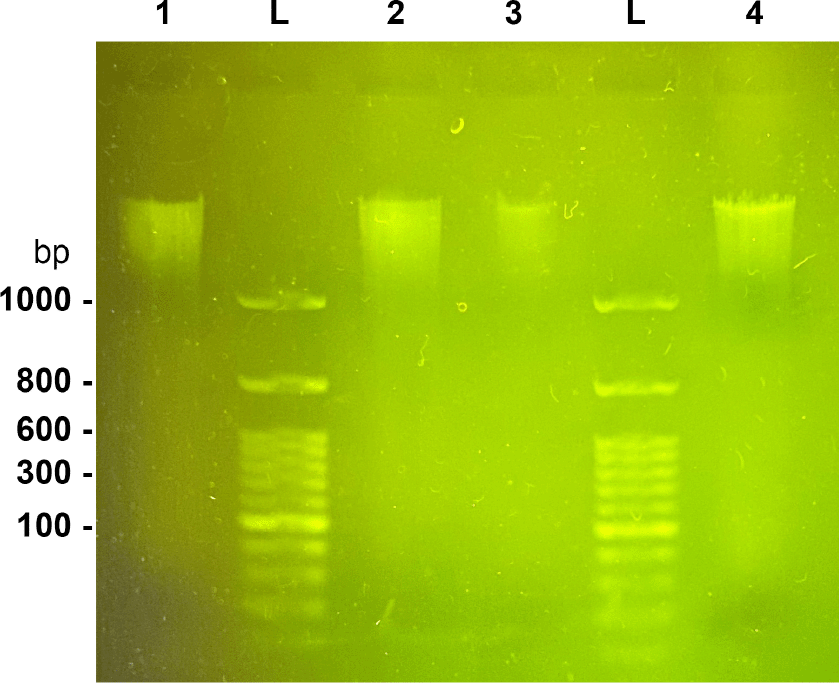
Sample 1 (5 l), 1 kb ladder, Sample 1 (10 l), 1 kb ladder, Sample 2 (5 l), 1 kb ladder, Sample 2 (10 l).
The tube was allowed to sit for 10 min at room temperature. 150 l of 100 mM Tris, Boric acid, EDTA (TBE) pH 8.3 buffer (Bioland Scientific, TBE01, CA) was added and the tube was incubated in a thermocycler heat block for 10 minutes at 95 °C. The tissue debris was then pelleted for five minutes at 5000 × g (10,000 rpm) leaving the genomic DNA used as the template for the PCR reaction left remaining in the supernatant.
The PCR reaction was prepared in a separate 0.2 ml microcentrifuge tube by mixing 9 l of sterile distilled water, 0.4 l of 10 M forward primer, 0.4 l of 10 M reverse primer, and 11 l of 2× Taq PCR Premix (Bioland Scientific, TP01-00, CA). Template DNA was added to the mix by drawing off 1 l of supernatant from the top of the tube used in the previous lysis step.
Common fungal DNA barcoding primers ITS1-F (5′-CTTGGTCATTTAGAGGAAGTAA-3′)11 and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′)12 were used to amplify the internal transcribed spacer (ITS) region of the fungal ribosomal encoding genes. The cycling conditions were as follows: an initial denaturation step (2 min at 94 °C), 35 cycles of denaturation (94 °C for 30 secs), annealing (55 °C for 2 min) and elongation (72 °C for 1 min), and a final elongation step (72 °C for 10 min).
Successful PCR amplification of the barcoding locus was confirmed via electrophoresis using the MiniPCR blueGel system (Amplyus, MA) operating at a fixed 48V.
A total of 5 l of PCR product was mixed with 1 l of 6× DNA loading buffer (Bioland Scientific, DSB01-01, CA) and loaded into a 1% TBE gel. The gel was run for approximately 30 minutes until a bright band was visible and a size of 400-700 bp approximated by comparison with a 50 bp DNA ladder (Bioland Scientific, DM02-02, CA).
A total of 15 l of PCR product was transferred to a new 0.2 ml microcentrifuge, tightly wrapped in parafilm and sent through USPS Priority mail in a small padded envelop for forward-read Sanger sequencing (MCLab, Oakland, CA).
The resulting chromatogram was analyzed, trimmed and error-corrected by hand using SnapGene Viewer (GSL Biotech LLC, CA). The resulting sequence was then searched against NCBI GenBank using BLAST and aligned against type and ex-type sequences.
Fungal tissue preparation
A double-layer media plate was prepared using a 9 mm Petri dish with a 30 ml Malt Extract Agar (MEA) bottom-layer and a 20 ml Potato Dextrose broth top-layer. The top liquid layer was aseptically inoculated with a sterile toothpick by transferring a small amount of conidia from a sporulating culture into the center of the broth and was gently agitated.
The plate was then covered and left at room temperature in ambient light for several days until a thick mat of fresh non-sporulating mycelium had completely covered the top layer of liquid media.
The entire mycelial mat was lifted off the plate and transferred to a flat surface using sanitized forceps and patted dry with a paper towel lightly sprayed with 70% isopropyl alcohol. Approximately 1 g of wet mycelium was cut, transferred to a 1.5 ml microcentrifuge tube and macerated with the blunt end of a plastic inoculating loop.
Genomic DNA extraction
DNA extraction was then carried out from the prepared tissue using the Quick-DNA HMW MagBead Kit (Zymo, D6060, CA) with the following modifications to the protocol:
• 200 l each of DNA Elution Buffer and Biofluid & Solid Tissue Buffer were used to accommodate the increased starting volume of solid tissue. There was no change to the proteinase K volume called for in the protocol.
• The sample was incubated for 24 hours in a water bath at 55 °C).
• The sample was macerated with a plastic micro-pestle to ensure good homogenization and tissue lysis after adding the initial buffers, midway through the initial incubation period, and finally prior to the initial centrifugation steps preceding DNA purification.
• The entire volume of supernatant was used to carry out the DNA purification steps and the volume of Quick-DNA MagBinding Buffer was increased to match the volume of supernatant used.
• DIY-friendly magbead seperator and vortex mixer were used to carry out the remaining steps of the DNA purification protocol.
DIY magnetic bead separator rack
A low-cost diy-friendly magnetic bead separator rack was constructed using packing tape, super strong neodymium disc magnets (32 mm diameter × 3 mm thick) and a cardboard box. A video demonstration is available on Figshare (Extended data31).
DIY vortex mixer
A vibrating back massager with a flat silicon head attachment was wrapped in a piece of rigid cardboard and affixed with tape to provide a contained area for the sample to vibrate. The massager was operated at low-speed for equivalent time periods in all steps requiring vortex mixing. While a lab-grade vortex mixer can be acquired at or below the cost of the massager used in this research, this device was already available to the researchers and further demonstrates the potential viability of alternative tools to those working in DIY lab environments. A video demonstration is available here (Extended data32).
The initial size and concentration of the extracted genomic DNA was estimated using gel electrophoresis with 1% TBE gel at a fixed 45 V and a 1 kb ladder (Bioland Scientific, DM01-01, CA).
The microcentrifuge tube containing the extracted DNA was tightly wrapped in parafilm, put in a padded envelop and shipped via USPS Priority mail from Los Angeles, California to Toronto, Canada where it arrived 11 days later.
Approximately 1 g was brought forward for library preparation. A DNA sequencing library was prepared with the SQK-LSK109 kit (Oxford Nanopore Technologies) according to the manufacturer’s protocol version GDE_9063_v109_revK_14Aug2019. Approximately 5 gigabases of sequencing data was collected using an R9.4.1 flow cell. Basecalling was performed with Guppy v5.015 in superior accuracy mode (Oxford Nanopore Technologies).
Sequencing quality was assessed using NanoPlot.13 All reads were used for assembly with Flye v2.914 using the parameters –nano-hq, –genome-size=42m, –threads=16. A 5 kb contig was removed because it was determined it was not part of the genome after performing a web-BLAST search. Polishing was performed with 1 round of Racon v1.4.2215 and one round of medaka v1.4.4 (Oxford Nanopore Technologies), both with default settings. The assembly was annotated using Liftoff v1.6.116 comparing against the Pestalotiopsis sp. NC0098 genome.17 Quality of the genome assembly was evaluated using BUSCO v5.2.2 using the sordiomycetes_odb10 database.18
To estimate the number of chromosomes, we used a previously described network-based approach.19 We identified the telomere repeat sequence as AACCCT from the initial assembly. All reads containing a minimum of three consecutive repeats of this sequence (or the reverse complement) were extracted using grep. Telomere-containing reads were aligned against each other in all-vs-all mode using minimap2.20 Alignments were then filtered to retain only alignments with more than 95% query coverage using awk. A network graph was generated using the iGraph R package.21 The workflow is available on Zenodo (Analysis code).
A list of type and ex-type sequences containing ITS, TUB and TEF accessions for Pestalotiopsis, including an out-group using the species Neopestalotiopsis saprophytica, was constructed from prior work.22 The resulting phylogenetic analysis is shown in Figure 4.
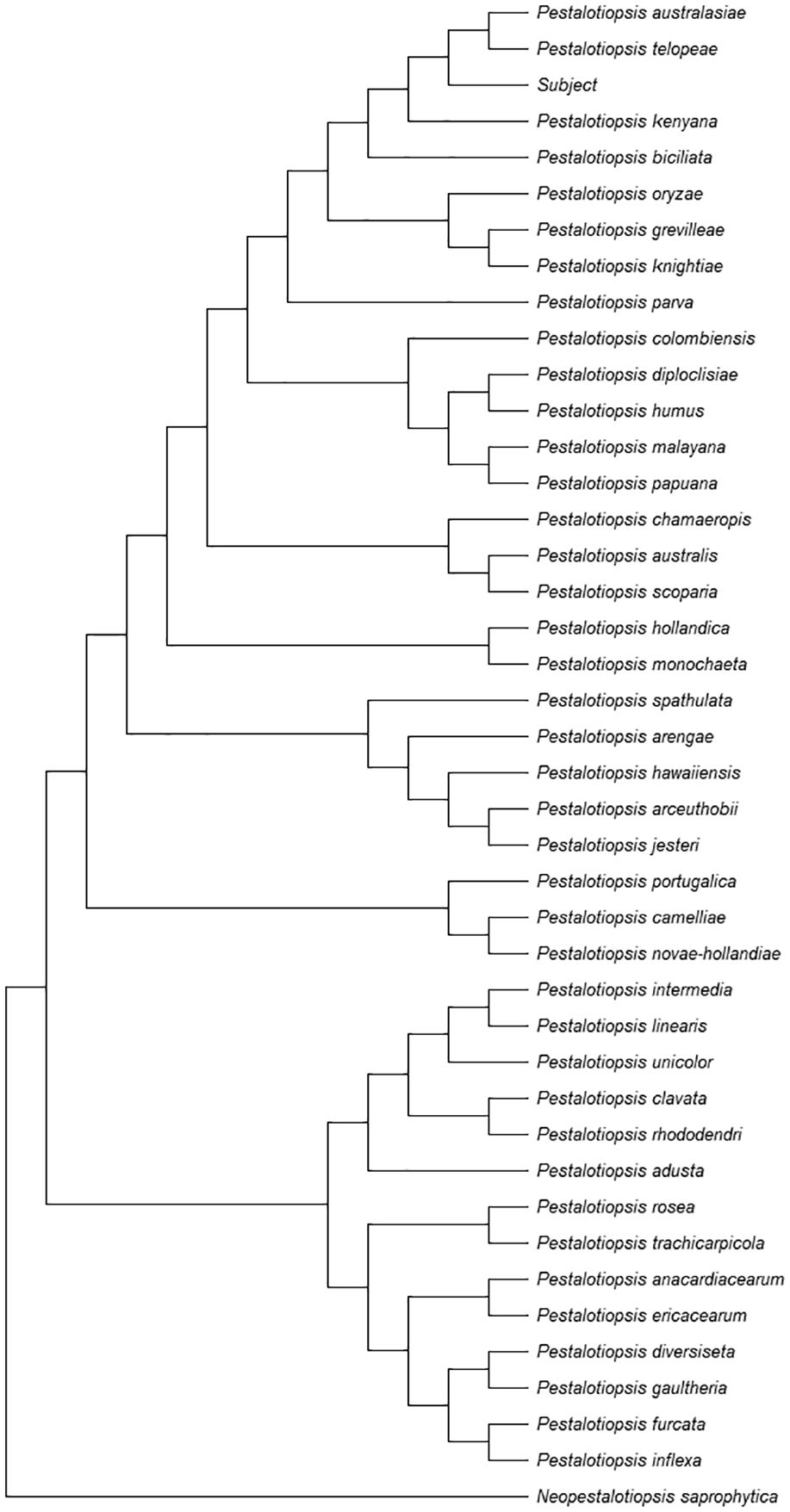
The collection of 42 accessions were aligned with MAFFT v7.45323 using the ‘–auto’ configuration command-line argument. The resulting alignments were trimmed and concatenated using Mega 11.24
A Maximum Likelihood (ML) tree was constructed in Mega 11 using the combined ITS+TUB+TEF alignment with default settings and 100 bootstrap replications.
The final tree was saved in Newick format and visualized by rooting the tree on the Neopestalotiopsis saprophytica outgroup.
The phylogenetic analysis workflow, scripts and data is available on GitHub (Extended data).
The majority of the DNA obtained from extraction was above 1 kb in length (Figure 3). After sequencing, we obtained a read N50 of 6.6 kb (Table 1), with approximately 105X coverage. Genome completion estimates revealed high completion, low duplication, and low fragmentation of the expected single-copy core genes. The total assembly length was 47.7 megabases, composed of five contigs with telomere repeats present at both the start and end of the contig, four contigs with no telomere repeats present, and a 68-kb circularized contig determined to be the full-length mitochondrial genome (Table 1). The BUSCO scores revealed high completion, with low missing and fragmented expected single-copy core genes.
BUSCO scores (v5.2.2) were calculated using the sordaiomycetes_odb10 lineage, revealing 97.9%C (S:97.5%, D:0.4%, F:0.6%, M:1.5%, n:3817).
After all sub-graphs were extracted from the network graph of telomere-containing reads, a total of 14 clusters containing telomere-containing reads were obtained (example of one is shown in Figure 5). Since each cluster represents one telomere and there are two telomeres per chromosome, this results in a total of seven linear nuclear chromosomes.
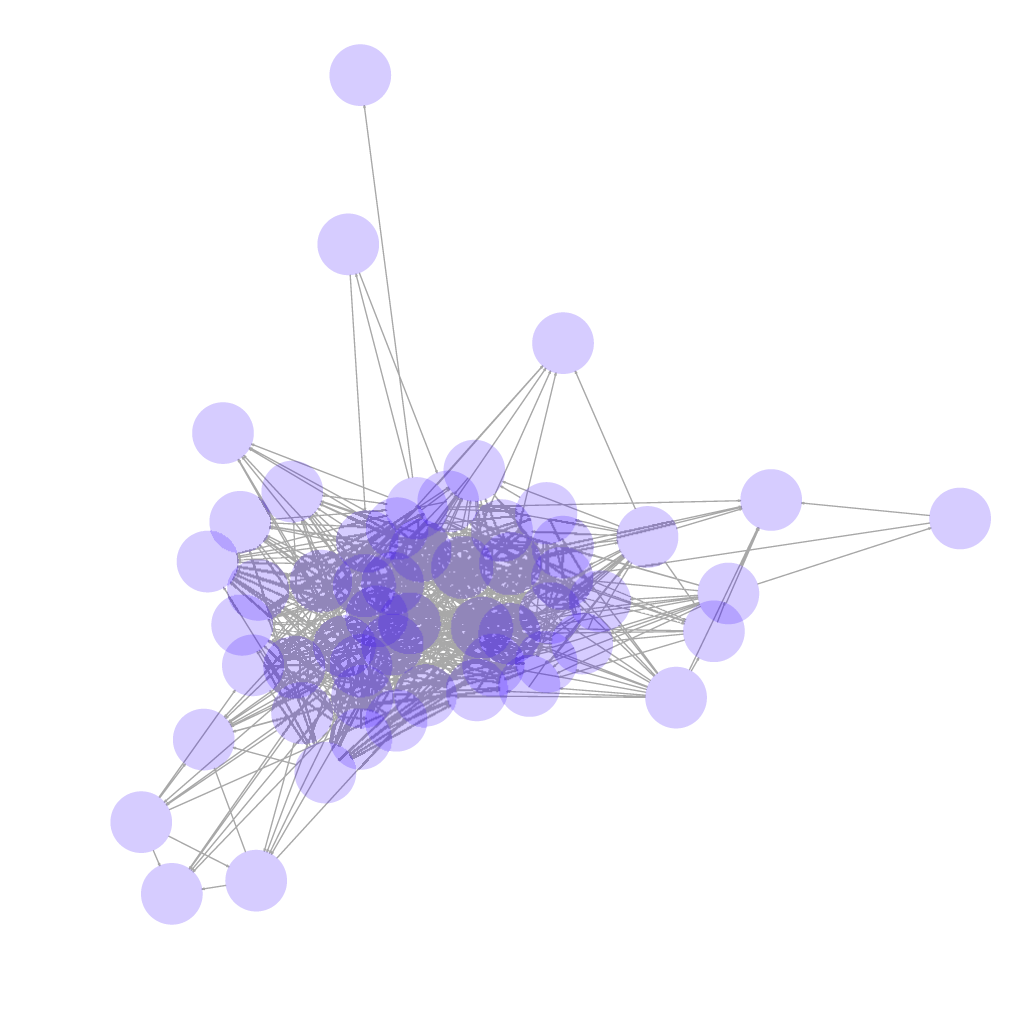
This network graph shows the network of long reads that are derived from a single telomere.
The cost for consumables for this project was determined to be approximately $300 on a per-sample basis (Table 2), however, the total upfront cost would be approximately $3098.
Flow cell cost assumes 1/6 (i.e., 5 Gb of 30 Gigabases) of the throughput capacity was used.
| Item | Total cost | Per sample |
|---|---|---|
| DNA extraction | $259 | $3 |
| LSK-SQK109 | $599 | $99 |
| R9.4.1 Flow Cell | $900 | $150 |
| NEB Enzymes | $920 | $38 |
| Magnetic beads | $300 | $7 |
| Pipette tips, reagents | $120 | $5 |
| Total | $3098 | $302 |
A BLASTn search on the ITS, TUB and TEF regions of the isolate were obtained.
The ITS region showed similarities to P. grevilleae (NR_147548) with 99.5% (600/603), P. kenyana (NR_147549) with 99.83% (596/597), P. oryzae (NR_147547) with 99.83% (593/594), P. pini (MT374681) with 997% (601/606), and P. knightiae (KM199310) with 99.01% (599/605).
The TUB region showed similarities to P. photinicola (KY047662) with 98.83% (506/512), P. australasiae (KM199499) with 99.8% (491/492), P. trachicarpicola (JQ845946) with 98.05% (503/513), P. telopeae (KM199500) with 100% (481/481), and P. rosea (JX399069) with 96.88% (496/512).
The TEF region showed similarities to P. kenyana (KM199395) with 99.87% (774/775), P. oryzae (KM199398) with 99.87% (752/753), P. grevilleae (KM199407) with 97.25% (777/799), P. biciliata (KM199399) with 97.94% (762/778), P. knightiae (KM199408) 97.32% (763/784).
Here, we obtained a high-quality genome assembly for Pestalatiopsis sp., comprising five contigs with telomeres at both ends and four contigs without telomere repeats. We also showed that obtaining fungal DNA suitable for Nanopore sequencing in a DIY lab environment is possible by modifying the protocol of a low-cost commercially available extraction kit. Genome sequencing is now becoming more accessible because of the relatively low capital requirements of the Oxford Nanopore MinION platform. In addition, the basecalling and consensus sequence quality are rapidly improving, resulting in high-quality, Nanopore-only genome assemblies for a variety of organisms.25,26
While genome assemblers only report on the number and size of contiguous DNA sequences assembled, it is left up to the user to infer further information such as number of chromosomes. We used a previously developed approach19 based on network graphs to determine that 14 unique telomeres were present in the sequencing data. The DNA was extracted during a point in the life cycle where the genome is expected to be haploid. We therefore reason that seven unique telomere-capped chromosomes exist in this species, in addition to the circular mitochondrial genome.
One known quality concern with nanopore-only genome assemblies is the presence of frame-shifting indels that can affect protein prediction.27 In this assembly, the high BUSCO completion score of 97.9% and low fragmentation score of the single-copy core genes (F:0.6%) over 3817 single-copy core genes suggest minimal frame-shifting indels were present after polishing. Until recently, it was often necessary to polish nanopore genome assemblies with high-quality Illumina reads using a tool like Pilon to obtain high-quality nanopore assemblies.28 However, recent improvements to basecalling models and polishing algorithms enable high-quality genome assemblies from nanopore reads only. This further improves accessibility to high-quality genome assemblies by removing the need to sequence using an expensive platform, typically only found in labs with a large budget or institutional support.
With the exception of the nanopore library prep and sequencing, all organism isolation and culture work, genomic DNA extraction and microscopy was performed in a residential DIY home lab using consumer-grade or second-hand equipment acquired from consumer-facing online vendors like Amazon.com and eBay.com. This includes pipettors, a water bath, a thermocycler, a microcentrifuge, and a self-built laminar flow hood constructed with plywood, a 24x24x12 HEPA filter and inline duct fan. The cost of sequencing reagents for this genome was around $300; however, many of the reagents require the purchase of six to 24 reactions. All the software used in this project is freely available, thanks to the academic and open-source bioinformatics community.14,15,20,29 Furthermore, all computation (except for basecalling) was performed on a consumer-grade laptop (Lenovo Thinkpad P14s) with 36 GB ram, AMD Ryzen 7 PRO 4750U processor (eight cores, 16 threads) and 1 TB NVMe SSD storage.
Pestalatiopsis sp. is known to possess diverse metabolic capabilities, including the production of potentially valuable secondary metabolites such as antitumor taxols,8 polyurethane-degrading hydrolases9 and compounds like alkaloids, polyketides, terpenoids, flavonoids, coumarins, xanthones, quinones, semiquinones, peptides, phenols, phenolic acids, and lactones.30 This additional genome provides a high quality genome for another species, and a workflow to generate low cost genomes to the community for future studies in the metabolic capabilities of this genus.
Here, we generated a high-quality genome assembly using nanopore sequencing for Pestalatiopsis sp., and conclude that this species possesses seven nuclear chromosomes in addition to a mitochondrial genome. We conclude that genome sequencing for novel fungi species can be performed in a DIY environment for approximately USD$300 on a per-sample basis.
The strain of Pestalotiopsis used in this research is available upon request from Josh McGinnis ([email protected]).
BioProject: High-quality genome assembly of a Pestalotiopsis fungi, https://identifiers.org/bioproject: PRJNA773800
Figshare: DIY Magnetic Bead Separator Rack, https://doi.org/10.6084/m9.figshare.19129856.v2.31
Figshare: Vibrating Back Massager as DIY Vortex Mixer, https://doi.org/10.6084/m9.figshare.19129862.v1.32
Data are available under the terms of the Commons Attribution 4.0 International license (CC-BY 4.0).
Workflows, supporting scripts and accessions used in the phylogenetic analysis are available on GitHub (see below).
Analysis code available from: https://github.com/EverymanBio/pestalotiopsis
Archived analysis code as at time of publication: https://doi.org/10.5281/zenodo.6028895
License: GNU General Public License, version 3 (GPL-3.0).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)