Keywords
Microtia, Genetic Mutation, isolated, syndromic, Genetic Diversity, FGF3, HOXA2, TCOF1
This article is included in the Genomics and Genetics gateway.
Microtia, Genetic Mutation, isolated, syndromic, Genetic Diversity, FGF3, HOXA2, TCOF1
We have updated, revised, and rearrange the sentences and references as suggested by reviewers. We have also updated the percentage of genetic involvement and added the limitation of the study.
See the authors' detailed response to the review by Walid D. Fakhouri
See the authors' detailed response to the review by Isabella Monlleó
See the authors' detailed response to the review by Miriam Erandi Reyna-Fabian
Microtia is a congenital malformation of the outer ears caused by improper embryonic development.1,2 It is distinguished by small, irregularly shaped external ears, and it can occur unilaterally or bilaterally.1 The prevalence of microtia varies among ethnic groups (0.83–17.4 per 10,000 births).2–4 Microtia occurs unilaterally in 80%–90% of cases and bilaterally in 10%–20% of cases.2,4 Boys are approximately twofold more likely than girls to have microtia, and the right–left bilateral ratio is typically 6:3:1.2,3 Microtia may occur as an isolated condition, or as part of a spectrum of anomalies or syndrome and approximately 20–60% of children with microtia have associated anomalies or an identifiable syndrome.2,4 Microtia is easily misdiagnosed during pregnancy.5,6 If pregnancy ultrasonography suggests microtia, the diagnosis is easily confirmed and diagnosed following birth based on physical examination.5,6
The etiology of microtia and the causes of its variations remain unknown although there is compelling evidence that environmental factors such as maternal sociodemographic variables, multiple gestation, diseases (gestational diabetes, cold-like syndrome), and related drug treatments such as isotretinoin use during pregnancy play roles, genetic factors are also believed to influence the embryonic development of microtia.7–9 Estimates of the prevalence of inherited microtia vary greatly, ranging from 3 to 34%.10 Although certain studies discovered various candidate genetic disorders for microtia1,9,11–104 there is no single specific genetic disorder that is certain and always be found in every patient with microtia.
Research on animals with isolated microtia as a prominent feature revealed mutations in homeobox A2 (HOXA2), sine oculis homeobox (SIX), eyes absent transcriptional coactivator and phosphatase (EYA), TBX1, IRF6, and CHUK.105 The most common genes that identified to be involved in microtia related syndromic were PLCB4 and GNAI3 for auriculo- condylar syndrome; TFAP2A for branchio-oculo-facial (BOF) syndrome; EYA106–108 SIX1 and SIX5 for branchio-oto-renal (BOR) syndrome; CHD7 (SEMA3E) for CHARGE syndrome; FRAS1, FREM2, and GRIP1 for Fraser syndrome; MLL2 and KDM6A for Kabuki syndrome; GDF6 for Klippel–Feil syndrome; fibroblast growth factor 3 (FGF3) for labyrinthine aplasia, microtia and microdontia (LAMM) syndrome; FGFR2, FGFR3, and FGF10 for lacrimo-auriculo-dento-digital syndrome; EFTUD2 for mandibulofacial dysostosis with microcephaly; ORC1, ORC4, ORC6, CDT1, and CDC6 for Meier–Gorlin syndrome; HOXA2 for microtia, hearing impairment, and cleft palate; DHODH for Miller syndrome; SF3B4 for Nager syndrome; H6 family homeobox 1 transcription factor gene (HMX1) for oculo-auricular syndrome (OVAS); SALL1 for Townes–Brocks syndrome; and Treacher–Collins–Franceschetti syndrome 1 (TCOF1), POL1RC, and POL1RDT for Treacher–Collins syndrome (TCS).2
FGF3 mutations are commonly found in LAMM syndrome.11,12 The FGF3 protein regulates a cascade of chemical processes inside cells by binding to its receptor, thereby signaling cells to undergo particular changes, such as proliferating or maturing to perform specialized activities.109 TCOF1 mutations can cause TCS in up to 78% of patients.110 HOXA2 encodes key developmental transcription factors of the second branchial arch, which contributes significantly to the development of the external and middle ear in embryonic development, and it was previously linked to autosomal recessive bilateral microtia.10,13
To determine what genetic factors play a role in microtia, we conducted a first systematic review to identify the most common genes in microtia development qualitatively. Hopefully, this will help improve our understanding of microtia, highlight the importance of genetic screening as a diagnostic technique for preparation of further management.
We have registered our protocol with the International Prospective Register of Systematic Reviews (PROSPERO, CRD42021287294 (25/10/21)). We have also screened PROSPERO for similar systematic reviews. No registered protocol reviewing the genetic factors of microtia was identified. The report of this systematic review was formulated according to the recommendations of the Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) statement.111
We performed an extensive and systematic search of all published studies related to genetic factors implicated in the development or outcome of microtia. Rather than focusing on a single disease, we aimed to provide systematic evidence on all types of microtia, including isolated microtia and syndromic microtia. First, the identified publications were assessed for relevance to the topic of interest using their titles and abstracts. The identified articles were then examined for any duplication using Mendeley. Then, the complete text of all screened papers was reviewed for the inclusion criteria, which were observational studies and case reports/series in the English language that assessed genetic factors in microtia. The exclusion criteria were duplications, reviews, non-English articles, animal studies, and articles in which sufficient details on genetic factors of microtia were not provided.
Three of the four authors (A.S., I.L.P., and R.P.) performed the search and study selection, which was supervised by the fourth author (C.D.K.W). We used six electronic bibliographic databases, PubMed, Web of Science, Science Direct, Proquest, Springerlink, and Clinicaltrials.gov, to conduct systematic searches from 1 to 31 October 2021. We checked Medline (PubMed) to identify controlled vocabulary Medical Subject Headings terms related to genetics and microtia. Searching strategies for PubMed are presented in Supplementary Table 1 (see Extended data112) and modified for other electronic databases.
Data extraction was conducted independently by three reviewers (A.S., I.L.P., and R.P.) through a standardized form. The methodological quality of studies in this systematic review was assessed using Joanna Briggs Institute (JBI) critical appraisal tools.113 We extracted data once all of the screening and selection steps had been completed. For both syndromic and isolated microtia, two different extraction forms were produced. The following data were extracted: first author’s surname, year of publication, country of origin, study design, sample population, sex, age, type of microtia, analysis method, affected genes, and mutations. Disagreements between the three reviewers were settled by discussion with the fourth reviewer (C.D.K.W.).
All supplementary files can be found in the Extended data.112
We discovered 3,530 articles after searching six electronic bibliographic databases. In total, 183 publications were removed because they were not available in English, they were animal studies, their full text was not available, or they were duplicated studies. Only 113 articles were eligible for more extensive evaluation after title and abstract screening. Only 98 papers were included in this study after a comprehensive full text analysis (Figure 1). The included studies were reviewed, utilizing a checklist questions form provided by JBI based on the studies’ methodology. Based on the JBI Tools for case reports, case series, and case controls, all publications involved were assessed as low-risk bias (Supplementary Tables 2–4).
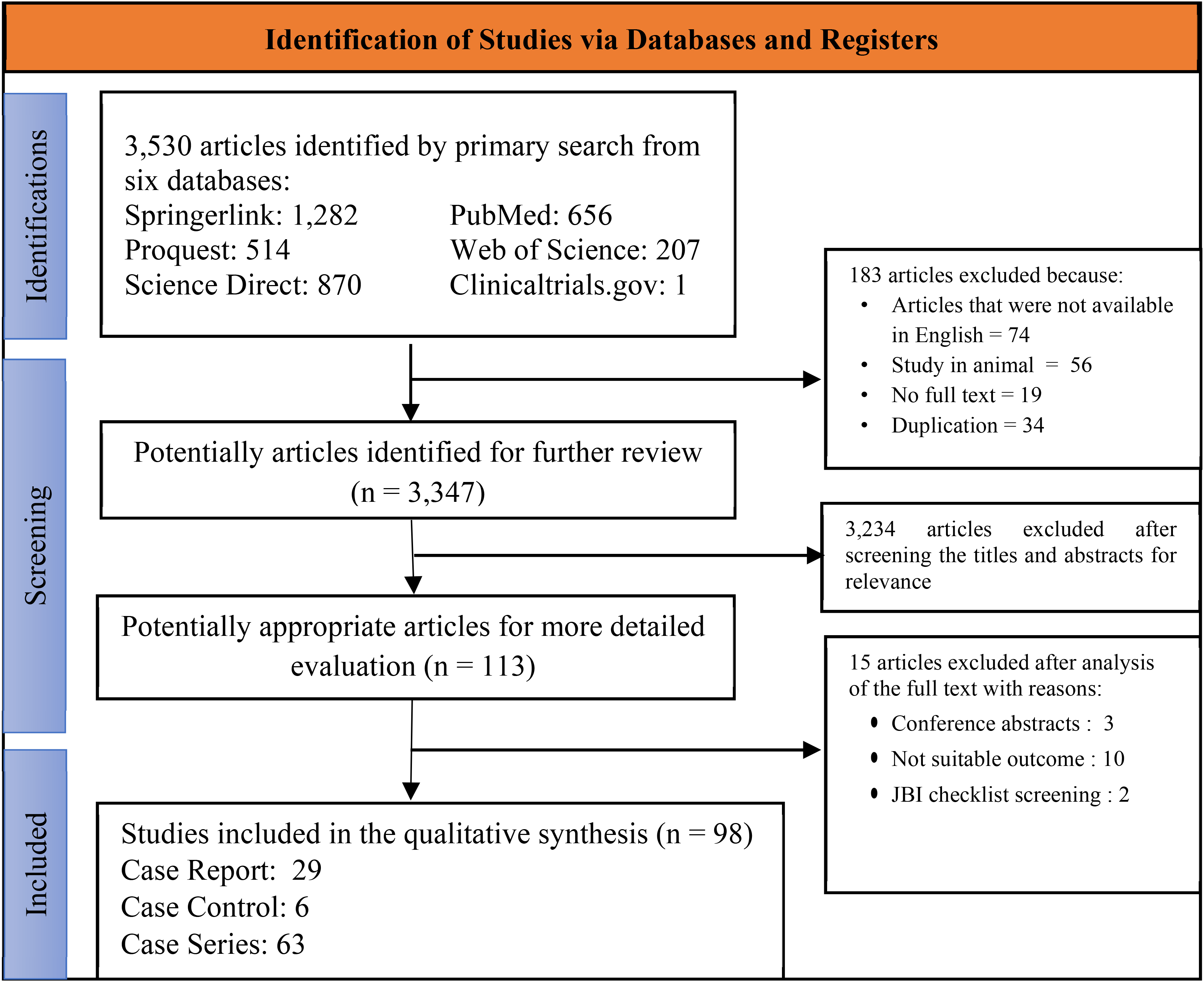
Following the screening, selection, and data extraction of all included studies, we discovered 79 articles of syndromic microtia involving 338 subjects (Table 1 and Supplementary Table 5) and 19 articles of isolated microtia involving 585 subjects (Table 2 and Supplementary Table 6). China had the most cases of syndromic microtia (58 subjects [17.1%] in 15 studies), followed by Italy (53 subjects [15.6%] in four studies); Turkey (36 subjects [10.6%] in seven studies); USA (31 subjects [9.2%] in 14 studies); Pakistan (27 subjects [8%] in one study); UK (23 subjects [6.8%] in three studies); Canada (21 subjects [6.2%] in two studies); Netherland (18 subjects [5.3%] in one study); German (14 subjects [4.1%] in four studies); Belgium (11 subjects [3.2%] in two studies); France (7 subjects [2%] in three studies); Taiwan and Poland (6 subjects [1.7%] in four studies each); Switzerland (5 subjects [1.4%] in three studies); Arab (4 subjects [1.2%] in one study) and India, Austria, Korea (4 subjects [1.2%] in two studies each]. China had the most cases of isolated microtia (520 subjects [88.8%] in 11 studies), followed by Turkey (38 subjects [6.5%] in two studies), the USA (21 subjects [3.5%] in four studies), Iran and Italy (3 subjects [0.5%] in one study each). China had the most cases of microtia among all investigations, being the site of 578 of 923 cases (62.6%). Concerning the study design, there were 29 case reports, 63 case series, and 6 case control studies included in this analysis.
Abbreviations: CNV, copy number variation; CS, case series; CR, case report; F, female; M, male; FGF3, fibroblast growth factor 3; FOXI3, forkhead box I3; PAX1, paired box 1; TCOF, Treacher–Collins syndrome; EFTUD, Elongation Factor Tu GTP Binding Domain Containing; HOXD, Homeobox D Cluster; MYT1, Myelin Transcription Factor; TWITST, Twist-related protein; STAG, Stromal Antigen; CNV, copy number variation; PMM, phosphomannomutase; SALL, Spalt Like Transcription Factor; GMNN, Geminin DNA Replication Inhibitor; MYO5C, Myosin VC; EYA, eyes absent transcriptional coactivator and phosphatase; SF3B2, splicing factor 3b subunit 2; SF3B4, Splicing factor 3B subunit 4; GSC, Goosecoid Homeobox; DONSON, DNA Replication Fork Stabilization Factor DONSON; GRIP1, Glutamate Receptor Interacting Protein 1; HSPA9, Heat Shock Protein Family A Member 9; POLR1A, RNA Polymerase I Subunit A; POLR1B, RNA Polymerase I Subunit B; RPS26, Ribosomal Protein S26; RPS28, Ribosomal Protein S28; TSR2, TSR2 Ribosome Maturation Factor; UBE3B, Ubiquitin Protein Ligase E3B; ZYG11B, Zyg-11 Family Member B; CDC6, Cell Division Cycle 6; CDC45, Cell Division Cycle 45; MCM5, Minichromosome Maintenance Complex Component 5; CDT1, Chromatin licensing and DNA replication factor 6; ORC1, Origin recognition complex subunit 1; ORC4, Origin recognition complex subunit 4; ORC6, Origin recognition complex subunit 6.
Abbreviations: CC, case control; CS, case series; CR, case report; M, male; F, female; CNV, copy number variation; CYP, cytochrome p450; eNOS, endothelial nitric oxide synthase; FGF3, fibroblast growth factor 3; GSC, goosecoid homeobox; HOXA, homeobox A; HMX, H6 family homeobox; IFN, interferon; IL, interleukin; PRKR, protein kinase R; SIX2, sine oculis homeobox 2; TGF, transforming growth factor; TNF, tumor necrosis factor; BMP5s, Bone morphogenetic proteins 5s; FGFR2, Fibroblast Growth Factor Receptor 2.
Regarding studies of syndromic microtia (Table 1), the family history was known for 299 of 338 subjects (88.5%), whereas the family history was not discussed in 39 subjects (11.5%). Of the 299 subjects, 190 (63.5%) had family histories of microtia. We also discovered that, of the 338 subjects, the gender was known for 284 subjects (84%), which included 156 males (54.9%). The severity of microtia was known for 403 of the 514 ears (78.4%) involved in this analysis. Of the 403 ears, 123 (30.5%) had grade I microtia, 202 (50.1%) had grade II microtia, 70 (17.3%) had grade III microtia, and 8 (1.9%) had grade IV microtia. The type of microtia was known in 233 of 338 subjects (68.9%); of these subjects, 31 (13.3%), 26 (11.1%), and 176 (75.5%) had right unilateral, left unilateral, and bilateral microtia, respectively. Based on the gene disorder levels in 79 studies including 338 subjects, 322 subjects (95.3%) in 70 studies had DNA-level disorders, and 16 subject (4.7 %) in 9 study had a chromosomal disorder. We discovered that of the 338 subjects, 107 subjects (31.6%) in 18 studies had TCOF1 gene mutations, whereas 55 subjects (16.7%) in 7 studies had FGF3 gene mutations.
According to studies of isolated microtia (Table 2), 573 of 585 subjects (97.9%) had a family history of microtia. The genders of the 585 subjects were known for 445 subjects (76%), which included 263 males (59.1%). The severity of microtia was known for 171 ears of the 639 ears (26.7%) involved in this analysis, among whom 11 (6.4%), 72 (42.1%), 88 (51.5%) had grades I, I, and III microtia, respectively. The type of microtia was specified for 87 of 585 subjects (14.8%), being right unilateral, left unilateral, and bilateral in 21 (24.1%), 12 (13.8%), and 54 subjects (62.1%), respectively. Regarding the genetic disorder level, of 585 subjects in 19 studies, 584 subjects (99.8%) in 18 articles had DNA-level disorders, and one subject (0.2%) had a chromosomal disorder. We discovered that 140 subjects (23.9%) in seven studies had mutations in the HOXA2 gene.
We discovered that of 923 subjects, the family history was known in 872 subjects (94.5%). In total, 463 of the 872 subjects (51.9%) had family histories of microtia. The genders of 729 of the 923 total subjects (78.9%) were known. Among the 729 subjects, 419 (57.4%) were male, and 310 (42.5%) were female. The severity of microtia was known in 574 of the 1.153 ears (49.7%) analyzed in this study. Among these ears, 134 (23.34%) had grade I microtia, 274 (47.7%) had grade II microtia, 158 (27.5%) had grade III microtia, and 8 (1.4%) had grade IV microtia. Based on the type of microtia, the type of microtia was known for 320 of 923 subjects (34.6%). Among these 320 subjects, 52 (16.25%) had right unilateral microtia, 38 (11.8%) had left unilateral microtia, and 230 (71.8%) had bilateral microtia. Based on the gene disorder levels described in all 98 studies, 909 subjects (98.5%) in 89 studies had DNA-level disorders, whereas 14 subjects (1.5%) in 9 studies had chromosomal disorders. We discovered that TCOF1 was the most common gene involved in syndromic microtia (66 subjects [19.52%]), followed by FGF3 (51 subjects [15.1%)], and HOXA2 was the most common gene involved in isolated microtia (22 subjects [40.2%]).
Based on the type of mutation (Table 3), we discovered that in syndromic microtia, the most common type of mutation in TCOF1 was deletion, being detected in 26 of 66 subjects (39.4%). In FGF3, the most common mutation types were missense, being present in 28 of 51 subjects (54.9%) each, and in isolated microtia, the most common mutation type in HOXA2 was nonsense mutation, being present in 16 of 22 subjects (72.7%).
Microtia is a congenital external ear deformity that can range in severity from minor anatomical problems to full ears absence (anotia). Microtia can be a single birth abnormality or part of a broader set of defects or syndrome.8 This systematic review attempted to describe the genes that play important roles in the development of syndromic and non-syndromic microtia. Only 98 studies on genetically linked microtia met our selection criteria. In this study, China had the highest number of microtia cases. The reported prevalence of microtia/anotia varies between 1 in 3000 to 1 in 20 000 births.1,2 The prevalence might vary by country and race/ethnicity but this is likely dependent on what forms of microtia are included in studies.116,117
We discovered that most patients with microtia had a family history of the disease, including 97.7% of patients with isolated microtia and 63.5% of patients with syndromic microtia. This is consistent with the existing literature, which describes Mendelian hereditary variants of microtia with an autosomal dominant or recessive mode of inheritance.10 The rates of familial microtia ranged from 3 to 34%.10,118
Based on the gender classification of each study, we discovered nearly 60% of all patients were male, including 54.9% of patients with syndromic microtia and 59.1% of patients with isolated microtia. In prior research, microtia was more common in male patients than in female patients, with a sex ratio of 1.5:1 and an estimated 20–40% greater risk in males than in females.1,4
Although microtia can arise bilaterally, 77–93% of patients have unilateral involvement.4 The most common form of syndromic microtia is bilateral microtia.1 Although bilateral microtia was more common in this study, the type of microtia was only known for 34.6% of subjects. Because of the inadequate data, this could represent a biased outcome for the most prevalent type of microtia.
Most published research on microtia reported the existence or absence of microtia and/or anotia with no further details on severity. Marx (1926), Weerda (1988), Roger (1977), Tanzer (1978), and Hunter (2009) all provided classifications of microtia.5 Their classification system was nearly identical, consisting of four grading levels.4,119 Most of the listed paper used in our study had same definition to grade microtia, Hunter classification. To homogenize the definition to grade microtia. In our study we use Hunter classification system for microtia when extracting data from 98 studies and data provided data in Tables 1 and 2. The most prevalent severity of microtia in our study was grade II, accounting for 47.7% of all cases, owing to the higher number of syndromic microtia cases than isolated microtia cases in our study that are commonly with a grade II microtia presentation.1,4
From all examined studies involved, the most common genes associated with microtia were HOXA2, FGF3, andTCOF1. We discovered that in syndromic microtia, the most common types of mutation were TCOF1 deletion (39.4%), and nonsense in FGF3 (54.9%), whereas in isolated microtia, the most common type of mutation in HOXA2 was nonsense mutation (72.7%). This was consistent with the literature, which indicated that the most common genes involved in microtia were HOXA2, FGF3, HOXD, ORC1, ORC4, ORC6, CDT1, CDC6, DHODH, HMX1, EYA1, and. TCOF1.105
FGF3 encodes a protein member of the FGF family.109 The FGF family has extensive mitogenic and cell survival activities and is involved in various biological processes such as embryonic development, morphogenesis, tissue repair, cell growth and inner ear formation in mice and chicken.109 FGF3 activates a cascade of chemical reactions inside cells that activate certain changes, such as dividing or maturing to take on specialized functions, by attaching to another protein known as a receptor.109 FGF3 haploinsufficiency could also be related with dental and hearing problems. FGF signaling is required for the appropriate development of the otic placode, a thickening of the ectoderm on the outer surface of a developing embryo from which the ear develops.120 FGF3 mutation is often found in syndromic microtia, mainly in LAMM syndrome. Congenital deafness with LAMM syndrome is an autosomal recessive disorder characterized by significant bilateral congenital sensorineural deafness coupled with inner ear defects, grade I bilateral microtia, and microdontia (small teeth) as its major phenotypic features.10,15 The finding of biallelic pathogenic mutations in FGF3 on molecular genetic testing confirms the diagnosis of LAMM syndrome in the proband.11,12,16,17
The TCOF1 gene, which encodes a suspected nucleolar phosphoprotein known as treacle, has been identified as the cause of TCS,10,121 an autosomal dominant craniofacial development disorder, in up to 78% of patients.18,110,122 Inhibition of mature RNA ribosomal (rRNA) production and gene transcription in neural folds prefusion during the early stage of embryogenesis may cause abnormal development due to treacle haploinsufficiency, caused by mutation in the TCOF1 gene, thus affecting proliferation and proper differentiation of these embryonic cells.123 To date, more than 50 mutations have been identified in the TCOF1 gene, most of which are insertions or deletions. TCS is characterized by cleft palate, hypoplasia of facial bones (particularly the mandible and zygomatic complex), downward slanting of palpebral fissures with colobomas of lower eyelids, external ear deformity, conductive hearing loss, and defects in brain development such as microcephaly and mental retardation.19–24 TCOF1 was the most prevalent gene found in this systematic review. This was because TCOF1 is a causal gene for TCS, which features microtia as one of its clinical symptoms.20 In addition to clinical findings, TCS diagnosis is also confirmed by detection of pathogenic variants of TCOF1, POLR1D, or POLR1B using molecular genetic testing, mainly inherited in an autosomal dominant pattern.124 In accordance with our result, TCOF1 gene deletions were reported to range from a single exon to a whole gene. Despite the fact that >97% of reported cases contained a pathogenic mutation identifiable by sequencing, Bowman et al. (2012) reported 5% of cases (5/92) with a big deletion, suggesting that the rate of large deletions may be higher than current data suggest.110
In Vertebrate, Hox genes are a subset of homeobox genes of which there are four cluster groups (A–D).1 Depending on the animal, there are four to 48 per genome of Hox genes.1 In human, there are 39 homeobox genes of the HOX family at four loci, HOXA, HOXB, HOXC, and HOXD on chromosomes 7, 17, 12, and 2, respectively.1 All Hox genes encode proteins that share a 60 amino acid domain called the homeodomain.1 In a variety of organisms, this structural motif is found in many different transcription regulators for a wide range of genes and plays important roles in cell differentiation.1 Homeobox genes mutation during development result in the transformation of different parts of the body.1 Homeodomain is a helix-turn-helix motif which is consist of three alpha-helices and an N-terminal arm.1 Site-specific DNA binding is achieved by interaction of the third helix with the major DNA groove.1 The N-terminal arm residues normally mediate contacts with the minor groove of DNA.1 Different pathogenic changes in the sequence of a homeodomain can affect stability and/or DNA-binding activity.1
Homeobox genes participate in the formation of the pharyngeal arches.4,25 They encode transcription factors that determine cell positional identity and morphogenesis during development, as well as switch on cascades of other genes that shared a 180 bp segment of DNA.1,4,25,125,126 The Hox gene family was discovered to be grouped inside the genome and ordered on the chromosome in the order of expression during development. This ordered pattern of gene expression could be part of a mechanism that generates morphogenetic specification.4,25 HoxA2, a member of the HOXA cluster, encodes a protein with a molecular weight of 41kD and is important in the regulation of development and morphogenesis in patterning the antero-posterior axis of the embryo of almost all metazoans such as in patterning and morphogenesis of the neural-crest-derived mesenchyme.
HOXA2 was discovered to be highly expressed in the second brachial arches (BA2), to express critical developmental transcription factors BA2, to play an important role in the development of the external and middle ear during embryonic development, and to be associated with autosomal recessive bilateral microtia as a member of the HOX gene family.10,13,25 In humans, abnormal or lost HOXA2 function, as well as early and late HOXA2 inactivation, results in auditory system malformations, primarily in the external and middle ear, such as a duplicated or absent auricle10,25 Consequently, HOXA2 has been proposed as a key transcriptional regulator of auricle morphogenesis.125 Individuals with a homozygous HOXA2 mutation have far more severe clinical symptoms than those with a heterozygous mutation. In a mouse model, inactivation of Hox2 early in development results in the absence of the pinna, whereas late inactivation results in a hypomorphic auricle.127
From the study of mice, we know that the HoxA2 protein is important for the development of the auditory system, mainly the outer and middle ear.1 The embryological origin of the inner ear is different than that of the middle ear and outer ear, which share a common origin.1 The inner ear is derived from an epidermal otic placode at the level of the hindbrain, whereas the middle and outer ears originate from the mesenchyme at the first and second pharyngeal or branchial arches.1 The formation of many craniofacial tissues is influenced by Hox genes.1 Hoxa2 is also a key gene for the facial somatosensory map.1 A significant percentage of microtic patients also present deficient facial components that originate from the same embryological structures.1
Aside from HOXA2, FGF3, and TCOF1, other genes linked to microtia include HMX1, POL1RC, POLR1D, GSC, SIX1, EYA1, SALL1, EFTUD2, SF384, FGFR2, GRIP1.105 Among these, HMX1 located on chromosome 4p16.1 is prominent. It is involved in the differentiation of the lateral facial mesenchyme downstream of embryonic patterning genes.9,128 In humans, duplication in the intergenic region downstream of HMX1 have been linked to OVAS, which is characterized by external ear and eye deformity.9 In a five-generation Chinese family with isolated bilateral microtia, a 10-Mb linkage locus covering 4p16 was discovered.9,26,128
We hypothesized that a link existed between the types of genes involved and the grade of microtia. For example, FGF3 deletions in LAMM syndrome have been clinically identified as grade I microtia.11,12,16,17 According to the MARX classification,10 the HOXA2 gene was common in the form of microtia type II and was exclusive to isolated microtia,1,13,14,26–29,125,127,128 and no specific type of microtia has been linked to the deletion of TCOF1.10,29 However this requires further studies to confirm these data.
The field of genetic variables in microtia research is still relatively extensive. More research on genetic variables that contribute to microtia is required, particularly using the next generation sequencing (NGS) and DNA microarray approach. NGS, massively parallel sequencing, or deep sequencing are all terms that refer to DNA sequencing technology that has transformed genomic research.29,129 In comparison to other technologies, NGS can sequence the entire human genome in a single day. Genomes may be examined without prejudice, allowing mosaic mutations to be detected.28 On the other hand, DNA microarray is a revolutionary technique for gene expression profiling.130 Microarrays were created as a method for mapping and sequencing vast amounts of DNA. DNA microarrays offer a far higher throughput and are less time consuming than previous approaches. By applying NGS and microarray for screening of genetic risk factors in microtia, the diagnosis of microtia could be made earlier and the patients could get a more comprehensive treatment.
This systematic review used recent available evidence and is the first systematic review to describe genetic factors in microtia. All studies included in this review were assessed as being of high quality. However, the limitations of this study included the heterogeneity of studies on the genetic evaluation of microtia in online databases, as well as the absence of information on the details of the subjects; thus, we could not perform a meta-analysis in this study. Gender information was unknown in 21% of patients, severity was unknown in 50.2% of subjects, and the type of microtia was unclear in 65.3% of patients. Case reports and case series were the most common study types in this systematic review. More observational research on genetic microtia is required to perform a more comprehensive systematic review and even meta-analysis. By applying NGS and microarrays to screen for genetic risk factors in microtia, it is possible to diagnose microtia earlier whether it belongs to isolated microtia or syndromic microtia. So that experts can provide education to parents regarding the diagnosis of microtia and further management plans that can be taken to treat these patients can be more thorough because they have been well prepared beforehand.
According to this study, most cases of microtia (62.6%) occurred in China, 51.9% of subjects had a family history of microtia, 57.4% of cases occurred in males, 71.8% of cases were bilateral, and 47.7% of cases were grade IImicrotia. From the studies involved, the three most common genes associated with the development of microtia were HOXA2 (40.2%), FGF3 (15.1%), and TCOF1 (19.52%). The most prevalent syndromes related to microtia were TCS and LAMM syndrome. Deletion mutations in TCOF1 were found in 26 patients (39.4%), missense were present in FGF3 in 28 patients (54.9%), and nonsense were present in HOXA2 in 16 patients (72.7%).
More research on genetic variables in microtia is required, particularly the use of NGS, massively parallel sequencing, or deep sequencing. We recommend that investigations on genetically associated microtia be conducted using observational studies, and the features of patients involved should be described more clearly and comprehensively in the future for better systematic reviews or even meta-analysis.
By understanding the three most dominant genes associated with microtia (HOXA2, FGF3, and TCOF1), we could promote the early screening and detection of microtia in the next generation, allowing us to provide better education and genetic counseling to patients with microtia regarding the possibility of microtia development in their children, and we hope that this systematic review will serve as a reference for the establishment of a global database of patients with microtia.
All data underlying the results are available as part of the article and no additional source data are required.
Harvard Dataverse: The Role of Genetic Factors in Microtia: A Systematic Review. https://doi.org/10.7910/DVN/4RRHH0.112
This project contains the following extended data:
• Supplementary Files.docx (Table 1. Medline (Pubmed) search strategy to identify published literature, Tables 2-4 Risk of bias evaluation of studies involved in this systematic review using JBI Checklist, Tables 5-6 additional characteristics of studies involved in this systematic review)
• Table Manuscript.docx (Tables 1-2 main characteristics of studies involved in this systematic review, Table 3 type of mutation found on analysis)
Harvard Dataverse: PRISMA checklist and flowchart for ‘The role of genetic factors in microtia: A systematic review’. https://doi.org/10.7910/DVN/4RRHH0.112
Data are available under the terms of the Creative Commons Zero “No rights reserved” data waiver (CC0 1.0 Public domain dedication).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)