Keywords
single cell transcriptomics, single cell proteomics, CyTOF, mass cytometry, single cell integration, CD11c B cells
This article is included in the Cell & Molecular Biology gateway.
single cell transcriptomics, single cell proteomics, CyTOF, mass cytometry, single cell integration, CD11c B cells
We have added a section in the Methods describing alternative options of the parameters that were explored for the integration steps (detailed in Supplementary Table 1 which was added to this version). We have revised the methods section for clarity and expanded the “Integration of datasets of unmatched samples from different conditions” section of the results considerably to explore best options for the integration of unmatched samples. Furthermore, in the same section, we have included an additional integration of all the twenty-four PBMC samples with the CyTOF samples of healthy controls from COMBAT (“refPBMC Mixed and CyTOF HC integration”) to further assess the accuracy of the integration using different reference datasets.
We also revised Figure 3 and added Table 2, five Supplementary Figures and a Supplementary Table to address the helpful comments of the Reviewer.
See the authors' detailed response to the review by Tallulah Andrews
See the authors' detailed response to the review by Xiang Chen
In the last decade the analysis of transcriptomes at single cell resolution has revolutionised molecular biology and become one of the most widely used techniques for answering fundamental biological questions of cell identities and functions.1–3 Single-cell RNA sequencing (scRNA-Seq) and mass cytometry (CyTOF) have both contributed immensely in the characterisation of cellular states, the identification of novel populations and the understanding of cellular differentiation trajectories.1,2,4–6 Protein and transcriptomic measurements in single cells, however, have been shown to provide different and complementary information regarding cell states.7,8 Joining the two fields can provide insightful observations on biological systems and functions at a cellular level.
The simultaneous quantification of transcriptome and proteins of the same cells has now marked the beginning of a new era in which our understanding of cellular systems and processes will be transformed by coupling the phenotypic and transcriptomics landscapes of biology. However, in these early days of multiplex technologies where cost and sample accessibility can be a limiting factor, there is still a lot to be gained from the integration of independent and already available datasets. Mass cytometry can target both intracellular epitopes and epigenetic markers with high precision and also has the advantage of being able to identify novel and very rare populations while profiling up to hundreds of thousands of cells per sample.7,9,10 Most importantly, the large quantity and high quality of single-modality single-cell data that have been already generated in the last few years have the potential to reveal novel biological insights when integrated appropriately.3,11 Therefore, the integration of CyTOF and scRNA-Seq can provide an affordable alternative to a unified view across modalities and aid in the functional characterisation of novel populations.
Here we present the multimodal integration of scRNA-Seq and CyTOF datasets coming from both matched samples (same patients profiled with the two platforms in the same dataset/study) and unmatched samples (different patients from distinct datasets/studies). We use the publicly available datasets from the multi-omic blood atlas of coronavirus disease 2019 (COVID-19) patients (COMBAT Study)12 to present the integration of matched datasets and show how the imputed values projected from the integration compare to the current gold standard measurements of ADT, coming from the available CITE-Seq experiment. Furthermore, we integrate the COMBAT mass cytometry dataset with an unmatched multimodal human PBMC atlas13 as an example of how scRNA-Seq and CyTOF datasets originating from different samples can also be integrated with high accuracy. We demonstrate that the integration of the CyTOF with the scRNA-Seq can guide the difficult task of annotating scRNA-Seq datasets and show that the imputed values generated are correlated to the CITE-seq protein levels. We finally use this integration technique to transcriptionally characterise a rare and heterogeneous subpopulation of CD11c+ B cells originally identified in the COMBAT mass cytometry dataset in a large meta-analysis of 12 COVID-19 scRNA-Seq datasets; thus, demonstrating the potential of this methodology to couple the unique strength of mass cytometry to identify very rare subpopulations with that of scRNA-Seq to functionally describe previously undefined subpopulations with unprecedented granularity.
COMBAT datasets
For the integration of matched datasets, we downloaded mass cytometry (CyTOF) and single-cell combined transcriptome (RNA) with surface proteome (ADT) data of peripheral blood mononuclear cells (PBMCs) from the Covid-19 Multi-omics Blood ATlas (COMBAT) Consortium12 (DOI 10.5281/zenodo.5139560). The datasets contained 91 overlapping samples from healthy controls and community COVID-19 cases in the recovery phase (never admitted to hospital) as well as from patients with mild, severe and critical COVID-19.
The scRNA-Seq and CyTOF datasets had been previously annotated, and the published annotations were retained as gold standard annotations. Both datasets were filtered to exclude cells with unclassified or uncertain annotations. Furthermore, subsets of cells that were extremely rare in the scRNA-Seq dataset (frequency less than 0.05%) and for which there was no overlapping features to identify them (namely reticulocyte and mast cells) were also removed prior to the integration. The scRNA-Seq and CITE-Seq datasets were further filtered to exclude low-quality cells based on i) number of genes, ii) number of features of ADT, iii) number of total UMI (RNA) or iv) number of total UMI of ADT lower than 0.001 of their relevant distributions. As this dataset is a subset of the published dataset (containing only the overlapping samples between scRNA-Seq and CyTOF), the raw RNA-Seq values were downloaded and re-integrated to mitigate batch effects. The batch correction was performed using the Seurat v3 Log normalize method with 50 PCA dimensions and 3000 highly variable genes and with the reciprocal PCA (RPCA) reduction for the anchor finding step.14 The batch correction was done on the 89 overlapping samples between scRNA-Seq and CyTOF (after the exclusion of 2 samples with less than 80 cells) on 182,000 cells by randomly subsampling 2000 cells from each sample and with the healthy controls as a reference. The published cell annotations which were created from expert immunological knowledge using data from all three modalities (GEX, ADT and VDJ)12 from the COMBAT Consortium were used for comparisons and the data was not re-clustered. The ADT dataset contained normalised values (normalisation using the DSB algorithm,15 details in Ahern et al.12) for 192 ADT tags, of which 39 had an equivalent CyTOF antibody.
The CyTOF dataset contained batch corrected and transformed values (arcsinh, with cofactor 5), for a panel of 45 markers (details for the batch correction can be found in Ahern et al.12). From those, 40 had an equivalent gene sequenced in the mRNA data (Table 1) and were used for the integration. The IgA and IgG antibodies had very poor staining in the mass cytometry experiment and were not included in any analysis. One channel included a combination of antibodies for IgD and TCRgd that was not used for the integration of the major cell types but was used for the integration of the B cells.
The gene names that correspond to the proteins from the mass cytometry (CyTOF) dataset and to the antibody derived tags (ADT) names from the COMBAT and refPBMC datasets.
For the B cell datasets, we extracted all cells from the scRNA-Seq that had been annotated as B cells (32,728 cells) and performed a batch correction with the healthy controls as a reference using Seurat v314 (89 samples after the exclusion of 2 samples with less than 80 cells). For the CyTOF dataset we subsampled 700 B cells per sample (57,159 cells).
Human PBMC Atlas datasets
The processed (after cellranger and QC) and annotated cell count matrices and the metadata of the scRNA-Seq was downloaded from the GEO database (GEO accession GS164378). The normalised and processed data object for the 3prime CITE-Seq dataset (ADT) was downloaded from https://atlas.fredhutch.org/data/nygc/multimodal/pbmc_multimodal.h5seurat (from here on named refPBMC). For the scRNA-Seq, each of the 24 samples were individually log normalised and scaled with linear regression of the covariate nUMI. To identify shared cell types across samples, batch correction and sample integration was performed for donor and time across 24 samples using the Seurat v3 Log normalize anchor based reciprocal PCA (RPCA) workflow with the unvaccinated samples (Day 0 samples) designated as the reference dataset, with 50 PCA dimensions and 3000 highly variable genes.
From the 161,764 cells, 146,480 cells were retained after filtering of cells annotated as doublets and those with less than 10 UMI counts for the common features between scRNA and CyTOF datasets. Additionally, the celltype.l2 annotations from the publication were renamed and grouped to best match the annotations from the COMBAT CyTOF dataset for visualisation and evaluation of the integration of the two modalities, but still included the cell-types exclusive to the refPBMC Atlas (erythroid cells, hematopoietic stem and progenitor cells-HSPCs, innate lymphoid cells-ILCs and platelets). The renamed annotations were: B (B naïve, B intermediate and B memory); CD4 (including all subclusters of CD4); CD8 (with all subclusters of CD8); dendritic cells (DCs) (ASDC, cDC1, cDC2, pDC); and NK (NK, NK Proliferating and NK CD56 Bright).
For the Day 0 integration, the refPBMC was filtered to include only the unvaccinated (Day 0) samples (47,962 cells after filtering of cells on the same thresholds as the full dataset). The unvaccinated samples were regressed for the nUMI covariate, batch corrected and integrated across the 8 donors using the same method and parameters as for the full dataset. The same process was followed for integrating the other two conditions (Day 2 and Day 7) separately.
COVID-19 datasets for the B cell CD11c+ subpopulation characterisation
For the characterisation of the B cell subpopulations, the count matrices, standardised metadata and the predicted celltypes were obtained from the processed HDF5 objects generated as part of the COVID-19 meta-analysis study carried out by Tian et al.16 (downloaded from https://atlas.fredhutch.org/fredhutch/covid/). The RNA count data matrices of all PBMC datasets were extracted for the cells which were predicted to be B cells for the predicted.celltype.l1 and we further removed cells annotated as Plasmablasts in the predicted.celltype.l2. Additionally, to exclude any non-B cells that might have erroneously been predicted as B cells in the original meta-analysis, only cells that had zero UMI counts of CD3E, GNLY, CD14, FCER1A, FCGR3A, LYZ, PPBP and CD8A genes were retained, as previously done by Stewart et al.17 After filtering for non-B cells, datasets with fewer than 60 cells were removed, leaving 13 datasets for downstream analysis.
The PBMC dataset of Su et al.18 was processed separately in order to use the ADT tags as validation of the CD11c+ subpopulation (25,443 cells). The dataset was split by batch and then individually log-normalised, scaled and linearly regressed for the covariates nUMI and sex. Then it was batch corrected with the Seurat v3 Log normalize anchor based workflow with 1000 common highly variable genes identified using the SelectIntegrationFeatures function and 15 CCA dimensions. UMAP dimensional reduction analysis was performed on the integrated object after scaling and regressing out sex with 15 PCA dimensions.
Each of the 12 remaining datasets was first individually log-normalised and scaled with linear regression for the covariates nUMI, sample and sex. The normalised and scaled dataset objects were then integrated and batch corrected with the Seurat v3 Log normalize anchor based workflow with 1000 common highly variable genes and 15 CCA dimensions. Subsequently, the integrated assay was filtered to only include samples from COVID-19 patients (77,485 cells) and it was rescaled and regressed for the covariate sex. UMAP dimensional reduction analysis was performed with 15 PCA dimensions on the batch corrected log normalised counts for downstream analyses.
For all analyses, the scRNA-Seq cells that had very low expression for all common protein-mRNA genes (less than 5 reads in total on all genes) were filtered out. The anchors between the query (scRNA-Seq) and reference (CyTOF) datasets were found based on a canonical correlation analysis (CCA) (with 20 dimensions for COMBAT and 25 dimensions for the reference PBMC dataset) using the FindTransferAnchors function from Seurat v314 (RRID:SCR_016341) and the defaults for the remaining parameters. Subsequently, the anchors were used to project the annotations of the CyTOF to the scRNA-Seq using the TransferData function with the integrated (batch corrected) PCA of the scRNA-Seq for the weight reduction and the CyTOF annotations as refdata. Equivalently, the TransferData function was also used to impute the CyTOF markers on the scRNA-Seq cells with the same parameters but using the CyTOF intensity values of the overlapping features as refdata. Finally, the imputed values were merged with the CyTOF data using the merge function and centered in order to run the UMAP19 and PAGA20 analyses to visualize all the cells together. The PAGA analyses were run using the scanpy toolkit (v1.9.1).21 We would like to highlight here that the ADT data is not used in any of the steps of the integration, but at the evaluation stage only.
Alternative options for both the anchor finding and data transferring steps of the integration were assessed at the initial stages of the analysis. These included: a) using the reciprocal PCA (rpca) reduction method in the FindTransferAnchors function (Analysis1); b) using the pcaproject reduction method in the FindTransferAnchors function (Analysis2); c) using the canonical correlation analysis (CCA) reduction as a weight reduction method for the TransferData function (Analysis3); d) using the pcaproject reduction method in the FindTransferAnchors function and the using the pcaproject reduction method in the TransferData function (Analysis4) and e) using the non-integrated (non-batch corrected) PCA of the scRNA-Seq for the weight reduction in the TransferData function (Analysis5). These options were tried independently keeping all the remaining parameters identical (apart from the ones listed above) and were evaluated using the Adjusted Rand Index (ARI) and F1 scores. Analysis1 had very similar results to the parameters tried above but all the remaining analyses were found to be suboptimal (Supplementary Table 1).
To assess the robustness of the integration for various cell numbers, the COMBAT scRNA-Seq and CyTOF datasets were downsampled to 9,100/22,750/45,500/91,000 and 182,000 cells for each modality by randomly subsampling 100/250/500/1000/2000 cells from each sample. The main results were presented for the dataset of 45,500 cells, as the most representative scenario of scRNA-Seq studies.
For the transcriptional characterisation of the B cell subpopulations, we integrated the CyTOF COMBAT dataset separately with 1) the scRNA-Seq COMBAT dataset, 2) the Su et al. PBMC scRNA-Seq dataset and 3) a meta-analysis of 12 PBMC COVID-19 scRNA-Seq datasets. The first two integrations served in finding a transcriptional signature for the CD11c+ cells, whereas the integration with the meta-analysis of the COVID-19 datasets was done to further define the heterogeneity within the CD11c+ subpopulation. For all integrations, a reduced set of 28 B cell and Plasma markers was utilised based on the features that were used to annotate these subpopulations in Ahern et al.12 The anchors and the projections of annotations and intensity values from the CyTOF were performed using a canonical correlation analysis (CCA) with 15 dimensions and k.weight=30 for the COMBAT and Su et al. integrations and k.weight=20 for the integration with the 12 COVID-19 scRNA-Seq datasets.
In order to evaluate how well the predicted annotations converged to the true cell types we used the Hubert-Arabie Adjusted Rand Index (ARI) for measuring the correspondence between two partitions of an object set22 and the F1 score. The ARI metric is adjusted for chance and random clusterings are expected to have an ARI equal to 0 and identical clusterings to 1. The metric compared the projected cell types from the CyTOF to the published cell annotations (including both common and dataset specific clusters) and was calculated using the implementation in the mclust R package23 (v5.4.9). The F1 score between the predicted and published annotations was calculated as the harmonic mean of precision and sensitivity for each cluster. For F1, the DN T cells and the cells for which there was not an equivalent annotation in both datasets were all grouped as Other. The cells from the cluster Other were retained to calculate the precision and sensitivity (misclassified cells) for each cluster but the F1 measure was not calculated for this cluster as it was a mix of multiple clusters. Finally, the F1 scores of two extra populations, the predicted Naïve CD4 and Naïve CD8 populations were calculated over T cells only.
To compare the imputed CyTOF intensities with the ADT normalised values at the pseudobulk level, we summarised the values (using the mean) on the pseudobulk clusters’ annotations (as annotated in the COMBAT study12) and the sample ID, using the function aggregateAcrossCells from the package scuttle24 (v 1.4). Then the values were correlated using Pearson’s correlation.
CD11c+ cells were determined using a threshold defined by the imputed protein values. We reasoned that the distribution of the imputed values for CD11c followed the CyTOF distribution of that marker since the imputation of the markers is a weighted average of the original values. Therefore, we firstly selected the CD11c+ populations of the CyTOF annotations. Then, for those cells, we chose the 1st quartile of the CD11c intensity distribution, which gave an intensity threshold of 1.6 (Supplementary Figure 1, Extended data42) to define the CD11c+ cells. Therefore, all cells with an imputed CD11c value greater than 1.6 in the Su et al. dataset were defined as CD11c+ cells. For the integration with the 12 COVID-19 datasets, the predicted annotations were used to define the subpopulations of interest (the cluster “CLA+ Switched memory B cells” comprised of 23 cells only and was removed from the downstream analysis). The transcriptional identity of the subpopulations was ascertained by finding DEGs using Seurat’s implementation of the Wilcoxon rank-sum test (FindMarkers default parameters, adjusted p value < 0.05). A gene set enrichment analysis (GSEA) was performed using the DGE results generated from FindMarkers (with parameters logfc.threshold=0 and min.pct = 0) and with the R package clusterProfiler25 (v 4.3.3) using the fgsea algorithm with the maximal size of genes annotated for testing set to 800 and minimum gene set size set to 10.
The R scripts used to run the analyses presented in this article are available from GitHub and archived with Zenodo.43
The ultimate motive of multimodal integrations is to take advantage of complimentary information of the datasets for a holistic understanding of cellular processes of interest. Stuart et al. demonstrated that transferring protein measurements across datasets can be a powerful tool to further our understanding of cell subpopulations. They also showed that this transfer can lead to accurate protein predictions even in the absence of a strong correlation between the RNA and protein of a gene, if alternative combinations of genes exhibit expression patterns that are correlated with cellular immunophenotypes.14 However, these integrations were performed based on RNA features alone. Here we extend this work to show that the integration of transcripts and proteins from RNA and CyTOF datasets based on mRNA-protein correspondences can also yield accurate predictions of cell populations and imputed protein features.
We used mass cytometry and CITE-Seq data from the COMBAT Consortium12 to showcase and validate the integration of the two modalities. The two datasets were downsampled to 45,500 cells for each modality (sampled as 500 cells per sample) to approximate a balanced real-life scenario and were integrated using Seurat with CyTOF as a reference and scRNA-Seq as a query with the Seurat v3 software.14 After finding the anchors between the two datasets, we used them to transfer the protein intensities to the RNA dataset, creating imputed values of the protein markers for each cell. We then merged the datasets to get a co-embedding of the two datasets on the same space (Figure 1A).
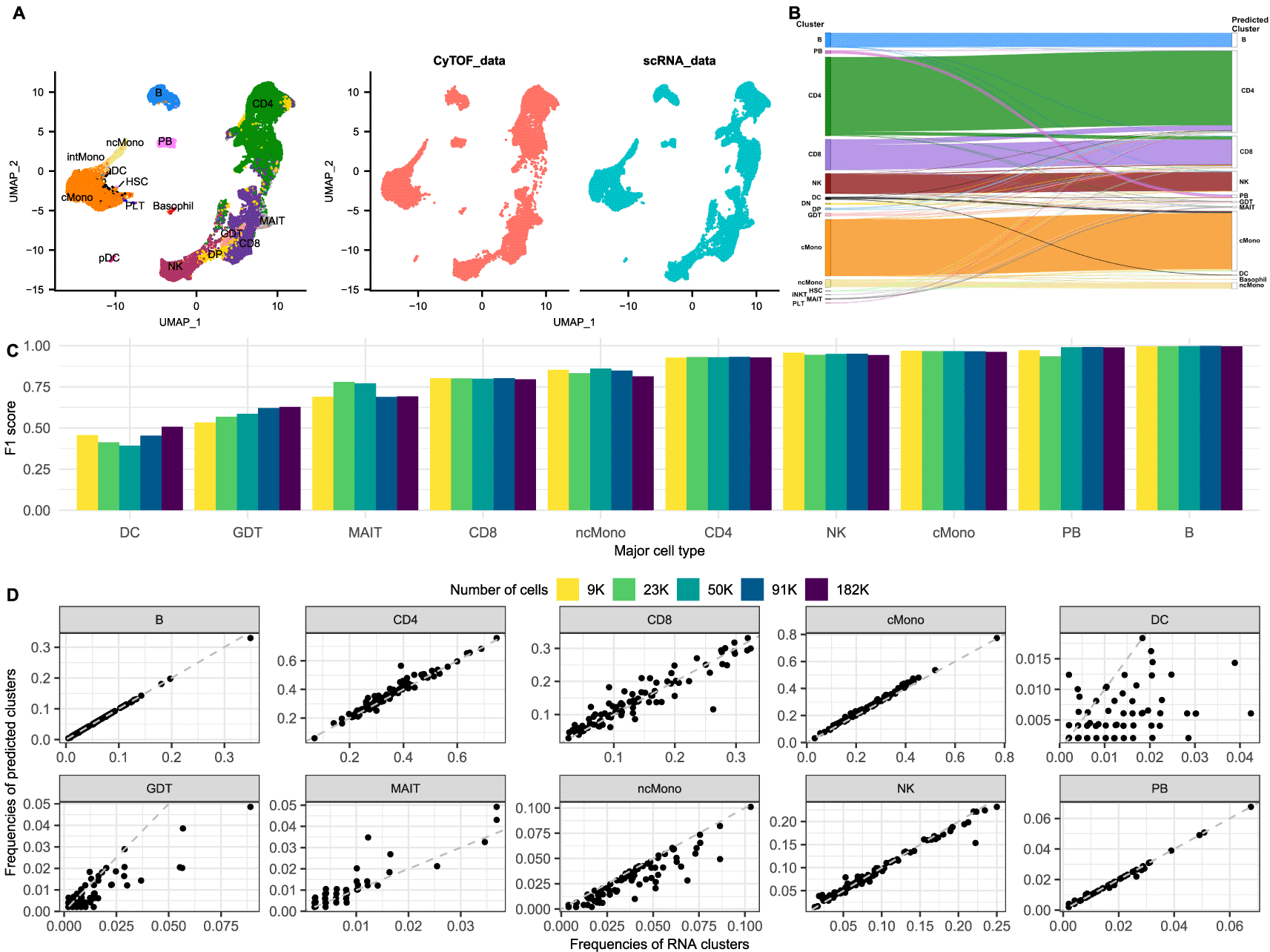
A. UMAP of the integrated COMBAT dataset coloured according to cell type (left) and split by modality (right). Red cells originate from the CyTOF (mass cytometry) dataset and the blue cells from the scRNA-Seq dataset, showing a good overlap between the two modalities.
B. Riverplot of the predicted labels (annotations) for each cell (right) compared to their actual major cell type (published annotations) in the COMBAT dataset (left).
C. Barplot of the F1 scores of the major cell types for different sample sizes of the COMBAT dataset. Each bar shows the F1 score for the integration using a downsampled subset of the data (number of cells according to colour).
D. Frequencies of the observed clusters (per sample cluster frequency based on published annotations) (x axes) compared to the frequencies as predicted by the integration from the CyTOF annotations (y axes) (per sample predicted cluster frequency) in the COMBAT dataset.
We observed that all major populations of the integrated dataset appear well separated in the uniform manifold and projection (UMAP) visualization and that all common populations are represented equally from both modalities (Figure 1A). Importantly, the T subpopulations are clearly distinguished, separating CD4, CD8 and NK cells. Interestingly, dataset specific subpopulations, such as the basophils from the CyTOF and the platelets from the RNA, form dataset-specific, distinct islands in the UMAP and are not mixed with other populations. The only populations that are not separating clearly and seem to be dispersed across the island of the monocytes are the conventional dendritic cells (cDCs) (cDC1 and cDC2 populations). To further validate the relationships of the cell types in a topologically meaningful embedding, we also generated the PAGA-initialized single-cell embeddings and PAGA graphs (Supplementary Figure 2) which confirmed the above observations. These results are in line with the findings in Hao et al. that CD8+ and CD4+ T cells were partially mixed when analysing the transcriptome but separated clearly when clustering on protein data, whereas cDCs formed distinct clusters when analysing RNA but were interspersed with other cell types based on surface protein abundance from the CITE-Seq dataset.13
One of the hardest tasks in the analysis of scRNA-Seq experiments is the annotation of cell types post clustering. Certain populations, such as CD4/CD8 T and CD8/NK cells are notoriously difficult to disentangle from the transcriptional data alone.13,26,27 Using the anchors provided by Seurat, the cell annotations of CyTOF can be projected to the scRNA dataset to inform the cluster identification of the latter. To validate the accuracy of these predictions, we compared the projected cell types from the CyTOF to the published cell annotations from the COMBAT Consortium which were created using expert immunological knowledge to guide a curated manual integration of the data from the different modalities (GEX, ADT and VDJ)12 (Figure 1B-D). Using the adjusted Rand Index (ARI) as measure of the similarity between two data annotations, we verified a high correspondence of the two label lists, with an ARI of 0.832. Furthermore, we calculated the F1 score of each population to assess the precision and sensitivity of the projections (Figure 1C). B cells and plasmablasts were the cell populations with the best scores, followed by classical monocytes, NK and CD4 T cells, all having a score above 0.9. Non classical monocytes, CD8 and MAIT cells had slightly reduced scores but were still well defined. On the other hand, 40% of gamma delta (γδ) T cells (GDT) were wrongly predicted as CD8 cells, due to the lack of common markers defining this population. TCRgd, the main marker for distinguishing GDT cells, could not be used for the integration because its antibody was added in the same channel as IgD for efficiency, and thus there was no one to one correspondence between the genes and the protein channels anymore. Furthermore, a large proportion of the DC cells were misclassified as classical monocytes, in line with previous observations that cDCs are well defined from transcriptomics signatures, but not easily separated based on surface protein abundance.13 Applying a filter for the prediction score (0.6) markedly improved the F1 scores of DC and GDT to 0.798 and 0.665, respectively, but reduced the number of cells with a prediction by 10.3%. These observations were further validated comparing the observed frequencies of the common cell types in the scRNA to the predicted annotations from CyTOF per sample (Figure 1D).
To quantify the amount by which the predictions are influenced by the number of cells in the two modalities, alternative scenarios were also examined. Smaller subsets were used to assess the robustness of the projections for lower numbers of cells and two scenarios of larger datasets were used to evaluate whether an increased number of cells further improves the predictions. Therefore, the analysis was repeated for 9,100, 22,750, 91,000 and 182,000 cells per modality. Most predictions remain consistently high even for the lower subsets of cells and increasing the number of cells does not seem to help all the clusters with low scores (Figure 1C). Finally, increasing the number of cells in the CyTOF dataset to 182,000 cells with the same resolution for the scRNA (45,500 cells) also does not provide a significant improvement in the scores (data not shown).
We next explored the potential of the transcriptomic and proteomic integration to reliably impute the protein markers on the scRNA data. The aim was to assess the accuracy of imputed markers for features that were used in the integration (common genes-proteins between RNA and CyTOF) and for features that did not have an equivalent gene, such as CD45RA and CD45RO. To evaluate the imputation, the transferred protein intensities were compared to the ADT values from the CITE-Seq data, which was considered the “ground truth” of protein measurements.
As expected, the accuracy of the imputation, measured using Pearson’s correlation of the imputed protein with the ADT, reflected to a certain degree the strength of the correlation between the RNA and real protein measurements (ADT) (Figure 2A). Genes for which the correlation between RNA and ADT was very low in the CITE-Seq data (below 0.2), did not provide a good proxy with the imputed protein values either. This included markers that were not relevant in the dataset (CD66 is a marker for neutrophils, of which there are none in this dataset) and antibodies that did not perform well in the ADT (CTLA4 and CCR7). Notable exceptions were CD141 (a marker for the cDC1 dendritic cells) and CD57 (a marker of cytotoxicity in T and NK populations), for which, even though the correlation between the transcriptome and the protein was poor, the integration was able to recover good proxies for the imputed features.
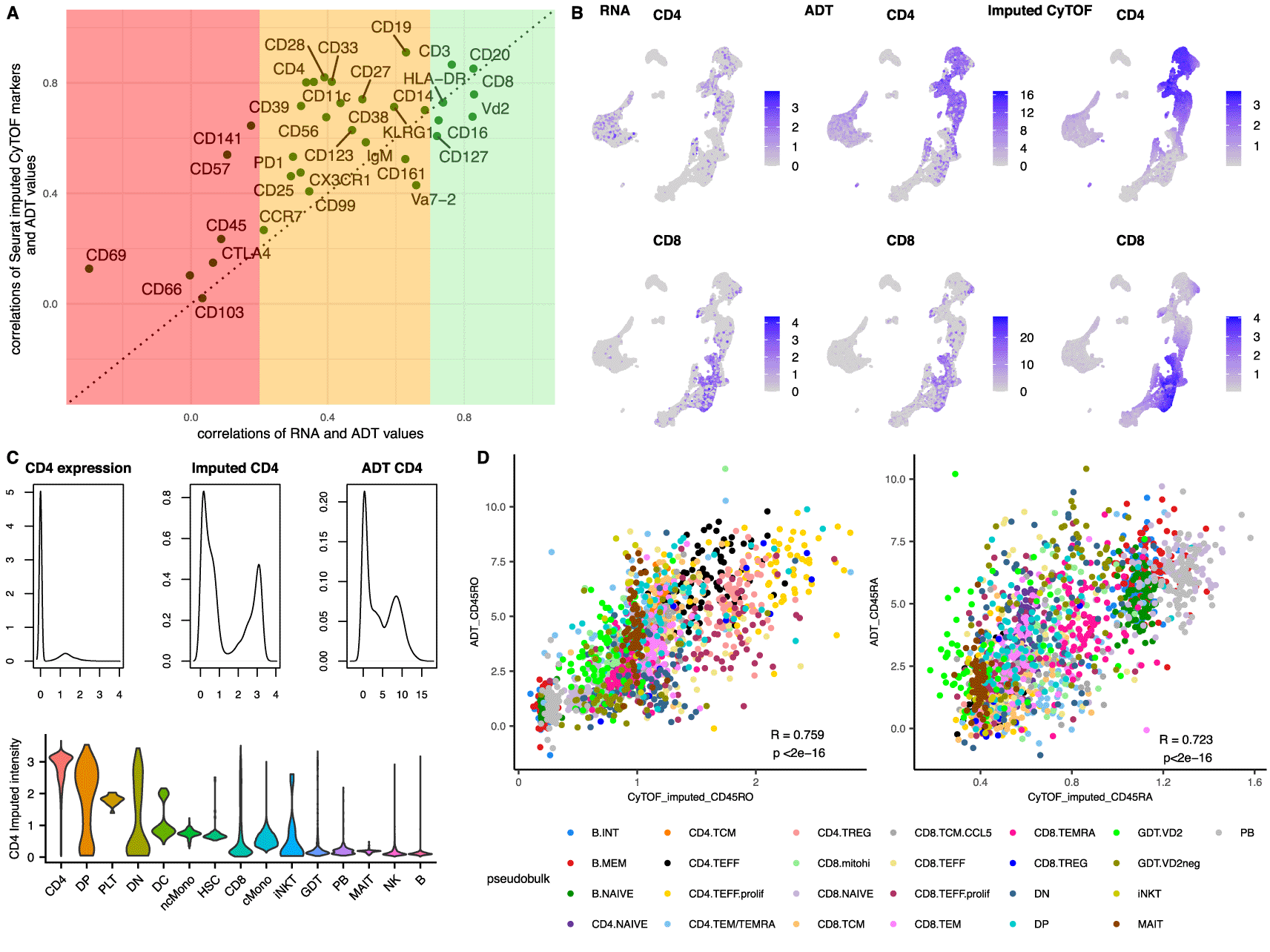
A. Scatterplot showing the correlation coefficients of the RNA to the ADT for each gene-protein pair (x axes) versus the correlations of the imputed CyTOF (mass cytometry) values to their equivalent ADT (y axes). The three coloured bands depict the strength of the correlations between RNA and ADT values of each gene-protein pair in regions of poor (Pearson’s correlations<0.2, red), medium (0.2<Pearson’s correlations<0.7, orange) and strong (Pearson’s correlations>0.7, green) correlations.
B. UMAP of the integrated dataset (constrained to the cells from the scRNA-Seq alone for all subpanels) showing the RNA expression (1st column) of CD4 (top row) and CD8 (bottom row) versus the ADT (2nd column) and the imputed CyTOF (3rd column).
C. Density plots of the CD4 expression (top 1st column), the CD4 imputed CyTOF intensity (top 2nd column) and the CD4 ADT (top 3rd column) followed by a violin plot of the CD4 imputed intensities according to the published annotations (bottom).
D. Scatterplots of pseudobulk populations per sample and cell type for CD45RO (left) and CD45RA (right). The averaged imputed value of the proteins is depicted on the x axes and the averaged observed ADT value for the same populations on the y axes.
Interestingly, most of the protein markers had a stronger correlation with their ADT counterpart than they did with their respective gene, highlighting that the anchor-based integration was able to recapitulate the protein intensities using the population structure similarities and not directly using the correlations of the proteins with the genes. As a result, we speculated that this integration can be used even for populations for which the correlations between the genes and the proteins is low to moderate. As additional evidence to this and to further assess whether this methodology could define populations which are transcriptionally challenging to disassociate, we focused on the CD4 and CD8 separation. As shown in Figure 2B–C, the imputed values of CD4 provide a very strong signal for the CD4 cells emphasising the differences between positive and negative populations and are strongly correlated with the ADT values (Pearson’s correlation 0.80), even though the gene is very lowly expressed and very poorly correlated with the protein values (Pearson’s correlations of expression to the ADT: 0.34 and to the imputed CyTOF: 0.24). In more detail, only 33.2% of the annotated CD4 cells had any expression of CD4 (more than 0 reads), whereas 90% of them had a high value for the imputed protein (imputed intensity > 2) suggesting that the imputed values could also be used to annotate scRNA-Seq datasets. Intriguingly, 12% of the CD8 cells also appeared to be CD4 positive using the imputed values, representing a CD8 subpopulation that was misaligned by the integration. Most of the CD8 cells with erroneously high imputed CD4 came from the CD8 T central memory (TCM) subpopulation, which has the lowest expression of CD8 amongst the CD8 subpopulations (Supplementary Figure 3, Extended data42). A differential gene expression between the two subpopulations (CD8.TCM vs CD4.TCM) revealed only 4 genes with a significant adjusted p-value and a logFC above 1 (CD8, CD8B, CD4 and CD161), suggesting a very similar transcriptional signature between the two populations, explaining this discordance.
Finally, we examined the correlations between the imputed CD45RA and CD45RO with their respective ADT. Even though these two markers were not used for the integration, weighted projected values could be calculated for each cell based on its anchors. Both CD45RO and CD45RA displayed a moderate Pearson correlation with the equivalent ADT (0.595 and 0.563 respectively, Supplementary Figure 4, Extended data42), but when the correlations were repeated on a pseudobulk level per subpopulation and sample, both markers exhibited a stronger correlation with the ADT, with coefficients of 0.759 and 0.723 respectively (Figure 2D), suggesting that even though the individual single cell predictions can be noisy, the bulk estimates are much more accurate. This was supported by the observation that the imputations were accentuating the differences between the positive and negative populations, creating a smoothing effect on the population imputed antibody intensities (Supplementary Figure 5, Extended data42). CD45RA and CD45RO are distinct isoforms of the CD45 protein and are encoded from the PTPRC gene. Following the same pattern as their ADT equivalent, neither imputed marker correlated with the PTPRC gene expression and they were shown to be mutually exclusive (Supplementary Figure 6). Furthermore, CD45RA and CD45RO are primarily markers that are used for the identification of naïve and memory populations of T cells. To provide additional evidence that the T cell subpopulation structure was captured correctly by the integration we calculated the F1 scores of the predicted Naïve CD4 and CD8 populations. Both populations had a high F1 score, with a value of 0.781 for Naïve CD4 and 0.801 for Naïve CD8. These observations further highlight the ability of the imputation to determine protein expression from the overall RNA profile of the cells rather than specific gene RNA expression patterns.
To further the utility of this method, we hypothesized that the integration could be used to inform transcriptomic datasets coming from unmatched samples and different conditions. The applicability of this would mean that publicly available CyTOF datasets of relevant samples could also help the identification of subpopulations in scRNA-Seq data of independent studies. To evaluate this type of integration, we used a publicly available dataset of twenty-four PBMC samples (hereafter named refPBMC) from eight volunteers collected at three time points: day 0 (immediately before); 3 days and 7 days after the administration of a VSV-vectored human immunodeficiency virus (HIV) vaccine,13,28,29 which we integrated with the CyTOF from the COMBAT Consortium12 (of healthy and COVID-19 samples) (from here on named “Mixed integration”). Consistent with our previous analysis, the integration successfully assigned the correct cell identities to 92.4% of the cells including all the cells which belonged to clusters that could not be resolved using the CyTOF information (erythrocytes, circulating innate lymphoid cells (ILC), HSPCs, platelets) and 92.7% of the cells excluding these populations of cells. The ARI between our predicted clusters and the published clusters (based on the WNN integration of RNA and ADT13) was 0.87, further confirming a good agreement between the annotations.
To evaluate if the main source of variability between the modalities is the immunological landscape of different individuals or the variety of conditions that these samples are coming from (COVID-19 infection vs HIV vaccination), we repeated the integration a) on the samples from day 0 (unvaccinated subjects only) with the CyTOF samples of healthy controls from COMBAT (in the following analysis named “Day 0 integration”) b) all the twenty-four PBMC samples with the CyTOF samples of healthy controls from COMBAT (in the following analysis named “refPBMC Mixed and CyTOF HC integration”). Qualitatively the three integrations are very distinct, where in the mixed integration the populations of B cells and monocytes form islands where the cells are coming from one modality only, whereas there is a much better overlap of cells in the unvaccinated and healthy controls (Day 0) integration for most major cell types (Figure 3A). This observation suggests that the variability observed in the integration of the mixed datasets is the result of biological variation between infection and vaccination effects, and not the result of the integration of different samples. Interestingly, most of the predicted annotations were similarly accurate in all three integrations apart from the plasmablast (PB) cluster (Figure 3B and Supplementary Figure 7, Extended data42). This subpopulation had an F1 score of 0.10 in the Mixed integration, whereas for the Day 0 integration the F1 was 0.83 and for the refPBMC Mixed and CyTOF HC integration it was 0.68. Ahern et al. showed an expanded PB population in COVID-19 patients indicating that abnormal frequencies in the reference population could create artefacts in the projected populations. In the Mixed integration, many monocytes were erroneously predicted to be PBs and had very low prediction scores (Supplementary Figure 8, Extended data42) highlighting that on integrations of unmatched datasets and different conditions, applying a prediction score threshold may be necessary to mitigate the effect of unbalanced population frequencies due to disease. Comparing the PAGA embeddings (Supplementary Figure 9), the Day 0 integration (COMBAT HC and unvaccinated refPBMC) shows the PB separating well and appearing the closest to the B cells, whereas in the Mixed integration (all COMBAT and refPBMC) the plasmablasts appear closer to the monocyte populations, highlighting the inaccuracies of the integration for this subpopulation. Contrary to what we would expect, for the Mixed integration we observed an equal representation of the misclassified PB on all samples, independent of day of collection (pre/post-vaccination) (Supplementary Figure 7, Extended data42) providing further support that the reference population was the cause of the inflated population of PBs.
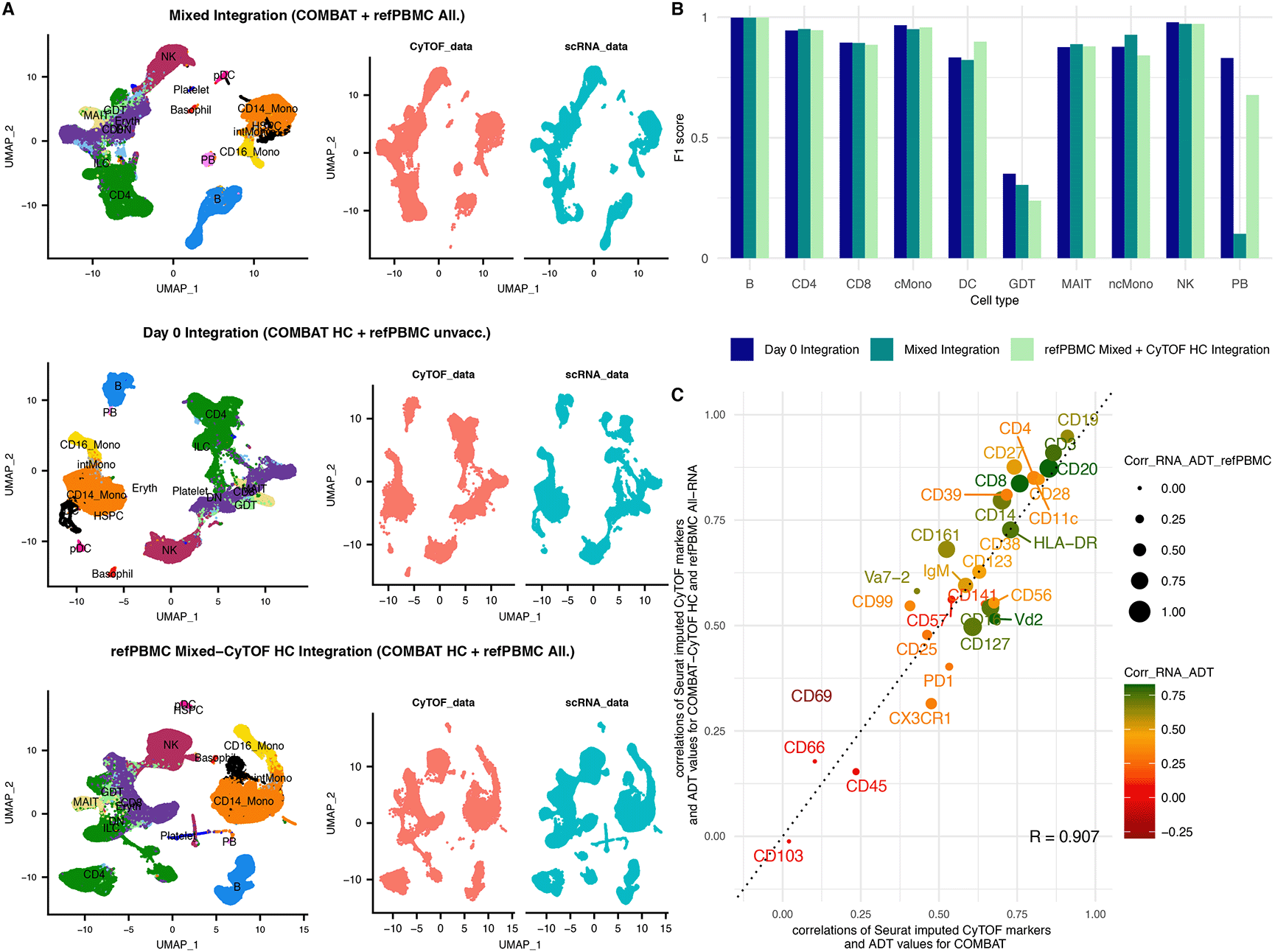
A. UMAP of the integrated dataset coloured according to cell type (left) and split by modality (right). Red cells originate from the CyTOF dataset, whereas blue cells from the scRNA-Seq dataset. The 1st row shows the mixed integration of the COMBAT CyTOF with the reference PBMC dataset (unvaccinated and vaccinated individuals); the 2nd row the Day 0 integration of the COMBAT CyTOF healthy samples with the refPBMC unvaccinated (healthy) samples only and the 3rd row the refPBMC Mixed-CyTOF HC integration of the COMBAT CyTOF healthy samples with all the refPBMC samples (unvaccinated and vaccinated individuals).
B. F1 score comparison for the Day 0 integration (dark blue); the Mixed integration (dark green) and the refPBMC Mixed-CyTOF HC integration (light green). All the integration scores are highly concordant for most celltypes apart from the plasmablasts (PB).
C. Scatterplot comparing the correlation coefficients between the imputed CyTOF and the ADT for each gene-protein pair in the COMBAT integration (x axes) versus the same correlations for the unmatched dataset (refPBMC Mixed-CyTOF HC integration) (y axes). The colour of the dot (and protein name) reflects the strength of the correlation of the RNA with the ADT values in the COMBAT dataset (red for poor correlation, orange for medium and green for strong correlation) and the size of the dot reflects the correlation of the RNA with the ADT values in the refPBMC dataset.
Having demonstrated the integration’s ability to accurately annotate the cell populations, we next imputed the CyTOF markers on the refPBMC dataset (refPBMC Mixed and CyTOF HC integration) to assess the granularity that these integrations can disentangle the underlying biological variation for unmatched datasets. To quantitatively validate the imputations, we calculated the correlations of the imputed protein intensities to the “ground truth” ADT measurements of the reference PBMC dataset. We noted a remarkable similarity of the correlations of the imputed markers with the refPBMC-ADT of the unmatched scenario to their respective correlations with the COMBAT-ADT of the matched integration (Figure 3C). Surprisingly, a number of imputed markers even outperformed their counterparts in the COMBAT integration, such as CD99, CD161 and CD14, suggesting differences in the efficiency of the staining of those antibodies between the two ADT datasets.
Finally, we proceeded to assess whether the batch correction of the RNA dataset influences the ability of the method to annotate rare subpopulations accurately. We compared the ARI and F1 scores of the “refPBMC Mixed and CyTOF HC integration” (in which the batch correction had been done on all samples together) with two alternative scenarios: a) of the COMBAT CyTOF healthy control samples being integrated separately with each individual sample of the refPBMC (per sample integration) and b) of the COMBAT CyTOF healthy control samples being integrated with each condition (Day 0/2/7) of the refPBMC separately (per condition integration) (Table 2). In each case the predicted labels of the per sample (or per condition) integration were concatenated to compare them to the published annotations. The ARIs from the three scenarios were very similar (ranging from 0.863 to 0.879) and showed little variation within each sample (Supplementary Figure 10). However, the F1 scores of the subpopulations DCs and the rare PBs showed considerable variation ranging from 0.594 to 0.899 for the DCs and from 0.569 to 0.829 for the PBs. For both populations the “per sample” integration had the lowest F1 score indicating the reduced power to detect rare or low frequency populations for small cell numbers. Interestingly, for PBs, the “per condition” integration showed a marked improvement in F1 score compared to the integration with all conditions together (0.829 vs 0.677). Comparing the correlations of the imputed CyTOF with the ADT values in the three scenarios, all markers apart from CD127 show an equal or higher correlation when all samples are integrated together compared to the per sample and per conditions integrations (Supplementary Figure 11). These results suggest no significant improvement of the integration when performed on a per sample or per condition basis, compared to the integration with the batch corrected dataset of all conditions together.
Table with the ARI and F1 scores for the three scenarios of integration: the CyTOF HC dataset integrated with each sample of the refPBMCs separately; the CyTOF HC with each condition (Day 0/2/7) of the refPBMC samples; and the CyTOF HC with all refPBMC samples together (refPBMC Mixed and CyTOF HC integration). The results reflect the ARI and F1 scores of the concatenated labels of all the samples together, not the average between all samples/conditions.
One of the most interesting applications of the integration of proteomic with transcriptomic datasets is the ability to use the former to identify subpopulations of interest and then characterise them in the latter, coupling phenotypic definitions to transcriptomic datasets. CD11c+ B cells are a heterogeneous subset of B cells (usually defined as CD19+, CD21- and CD27-) which is present at low frequency in healthy individuals30,31 but is increased in several conditions including malaria, HIV and autoimmune diseases.31–35 These cells have assumed many names in the literature, including ‘atypical (memory) B cells’ (or atBC), ‘Age associated cells’ (ABCs), ‘memory precursors’, ‘exhausted memory cells’31–33,36 and more recently have been subdivided into DN2 (double negative 2/extrafollicular ASC precursor B cells) and activated Naïve cells32 but there is no general consensus as to their definition. In the COMBAT Consortium, the authors reported subpopulations of the CD11c+ B cells that were significantly more abundant in critical COVID patients compared to healthy volunteers.12 Atypical B cells along with DN2 and activated naïve CD11c+ cells have also been shown to be increased in COVID-19,37–39 but their transcriptional heterogeneity after COVID-19 infection has not been explored. We therefore set off to find and transcriptionally characterise these cells in the scRNA-Seq dataset.
Using only the B cells subpopulations from both COMBAT CyTOF and scRNA-Seq datasets (excluding the plasmablasts), we were able to integrate the two modalities using as common features the markers that were selected for the B cells subclustering analysis from the COMBAT Consortium.12 In order to validate the results of the integration, we then downloaded the RNA and CITE-Seq data from an additional dataset from Su et al. of healthy and COVID19 samples18 and repeated the integration using the Su scRNA-Seq and the COMBAT CyTOF data. Even though both scRNA-Seq datasets had a low gene expression of CD11c, both independent integrations revealed a strong presence of CD11c+ subpopulations as defined by the imputed protein marker, which was also validated using the ADT values of the two studies (Figure 4A).
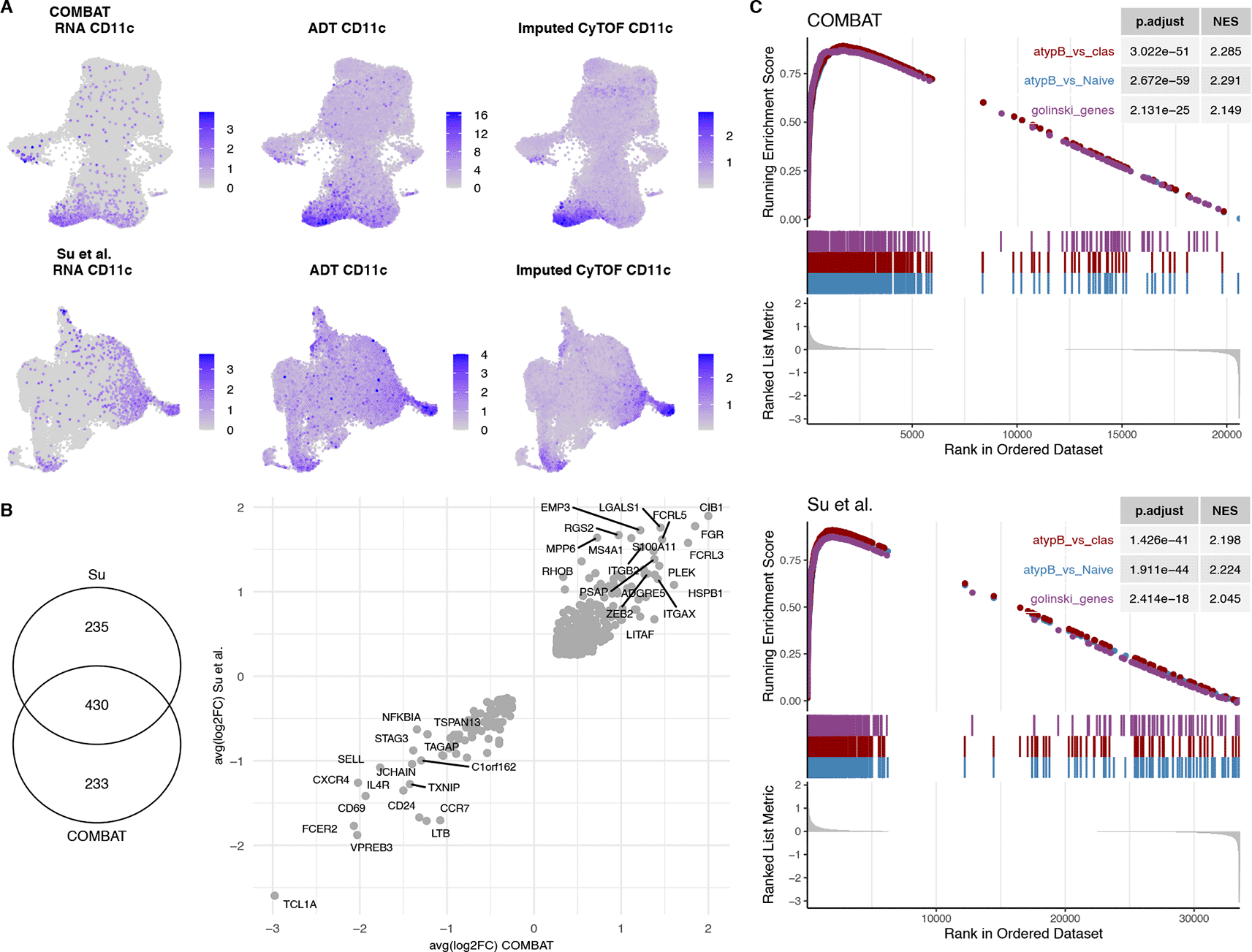
A. UMAP of the COMBAT scRNA-Seq (1st row) and Su et al. scRNA-Seq (2nd row) datasets showing the RNA expression of CD11c (1st column) versus the ADT (2nd column) and the imputed CyTOF (mass cytometry) (3rd column) validating the imputed CD11c predictions.
B. Venn diagram of the differential gene expression of CD11c+ vs CD11c- B cells for the COMBAT and Su et al. datasets. The average logFCs of the differentially expressed genes in both datasets (430 genes) are plotted as a scatterplot.
C. GSEA of differentially expressed CD11c+ genes from COMBAT and Su et al. datasets with two published gene list signatures of atypical memory B cells (MBCs) vs Naïve and vs Classical B cells of malaria infected individuals (Portugal et al.) and of CD11c+ B cells vs CD11c- from healthy donors (Golinksi et al.) showing significant enrichment of these gene sets.
Due to the low frequencies of the CD11c+ subpopulations, the naïve-like CD11c+ and the switched memory CD11c+ subpopulation could only be found in a fraction of the samples in both COMBAT and Su et al. datasets (Supplementary Figure 12, Extended data42). Therefore, we used the imputed values to identify the CD11c+ population of interest (details in the Methods section). Differential gene expression of the CD11c+ cells uncovered a transcriptional signature that had 430 genes in common between the two datasets (99 downregulated and 331 upregulated), all of which had the same direction of effects (Figure 4B). Moreover, we performed a gene set enrichment analysis (GSEA) of the two lists of differentially expressed genes from the COMBAT and Su et al. datasets with two published signatures of CD11c+ cells from healthy controls30 and from atypical BCs of malaria patients.34 Both show a significant enrichment for the CD11c+/atBC signatures (NES>2 and p-values<10-17), further confirming the identity of those cells (Figure 4C).
Amongst the genes with the largest logFC in both studies were FGR, CIB1, EMP3, MPP6 and RHOB (Figure 4B), genes that have been previously associated with the DN2 phenotype.17 Moreover, DN2 cells are known to express high levels of inhibitory receptors, such as those belonging to the family of Fc-receptor-like (FCRL) molecules,17,40 consistent with the upregulation of FCRL3 and FCRL5 in the CD11c+ subpopulation. TCL1A, on the other hand, is a marker of transitional and naïve cells, which is known to be absent in the other B cell populations.17 T-bet (TBX21) is a transcription factor that has been shown to be expressed in CD11c+ B cells, although not all CD11c+ B cells are T-bet+.30 In the COMBAT dataset TBX21 (T-bet) was filtered out due to low expression (expressed in less than 10% of the cells in both subsets) and could not be tested for differential expression between the two subsets. In contrast, TBX21 was significantly differentially expressed between the CD11c+ and CD11c- populations in the Su et al. dataset with a logFC of 0.51 (adjusted p-value<10-14). Additionally, many known transcriptional markers correlating with T-bet expression in B cells, such as ACTB, ALOX5AP, GSTP1 and LAPTM5,17 were enriched in CD11c+ cells for both datasets (Supplementary Figure 13, Extended data42).
To further explore the transcriptional heterogeneity of the CD11c+ population in COVID-19 patients, we integrated the CyTOF dataset with B cell populations of a meta-analysis of 12 scRNA-Seq datasets of COVID-19 studies.16 The analysis recovered all three CD11c+ subpopulations that had been found in the CyTOF COMBAT dataset. It identified 153 naïve CD11c+ (activated naïve cells/naive-like CD11c B cells, defined as CD27-, IgD+, CD11c+); 400 DN CD11c+ (DN2, defined as CD27-, IgD-, CD11c+) and a less well characterised population of 440 switched memory CD11c+ cells (CD27+, IgD-, CD11c+ cells). All three subpopulations, had a clear signature of genes associating with the CD11c+ phenotype, such as a higher expression of CD19 and CD20 (MSA4A1) as well as CD11c+ hallmark cell surface receptors CD84, CD68, CD8630; inhibitory receptors CD72, FCGR2B, FCRL3, FCRL5, LILRB133 and transcription factors TBX21, ZEB2 and ZBTB3234,35 (Supplementary Figure 14, Extended data42).
Pairwise analysis of the 3 subpopulations uncovered 91 differentially expressed genes between the DN CD11c+ cells and the switched memory CD11c+; 44 between the naïve-like CD11c+ and the switched memory CD11c+ (Figure 5A) and a very similar transcriptional profile for the DN2 and the naïve-like CD11c+ cells (no significant differentially expressed genes between the two), in line with the phenotypic similarities of these two populations that have been previously reported.32 The similarity between naïve-like and DN2 CD11c+ cells, and the differences between naïve-like CD11c+ cells and the naïve (CD11c-) cells (529 differentially expressed genes - data not shown), indicates that these naïve-like CD11c+ (or activated naïve) cells are transcriptionally closer to the DN2 than they are to the naïve counterparts. This suggests that these might not be naïve cells, but rather a small subset of CD11c+ cells sharing a limited set of phenotypic markers with classical naïve cells.
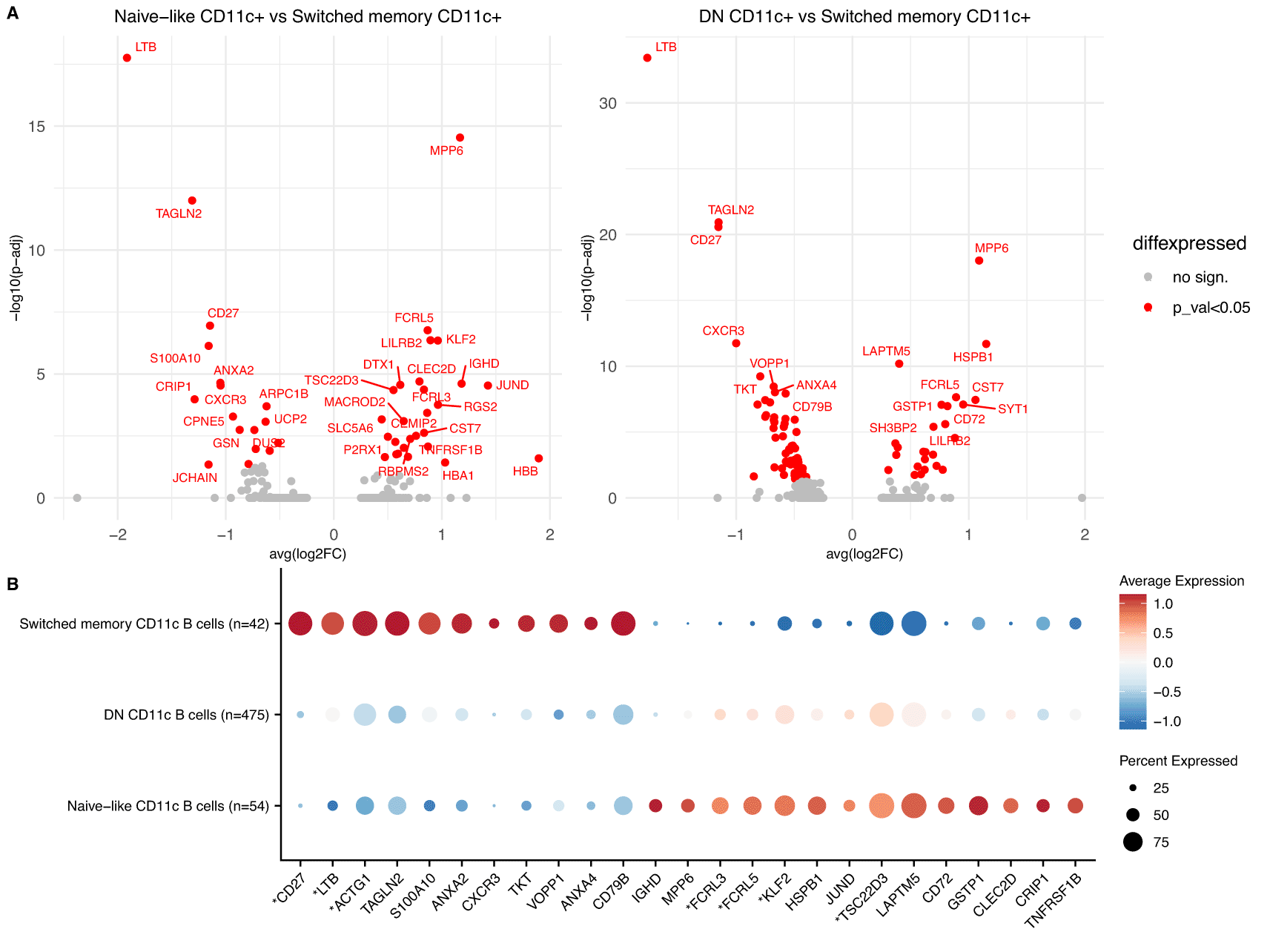
A. Volcano plots of DGE between Naïve-like CD11c+ vs Switched memory CD11c+ (left) and DN CD11c+ and Switched memory CD11c+ cells (right) in the meta-analysis of the 12 COVID-19 scRNA-Seq datasets. Significant genes shown in red.
B. Dotplot showing the top genes from A. for the 3 predicted subpopulations of CD11c+ cells in the dataset of Su et al. The genes with an asterisk were also shown to be significant in the comparison of Naïve-like CD11c+ cells vs Switched memory CD11c+ (p-adjusted<0.05).
The differentially expressed genes between the three subsets uncovered a subset specific expression of CXCR3, which was upregulated in the switched memory subset of CD11c+ cells (Figure 5A). CXCR3, an inflammatory chemokine receptor, has been shown to be upregulated in CD11c+ cells in multiple studies,31,38,40,41 with heterogeneous expression amongst these cells,38,40 but had not previously been associated with a specific subset of CD11c+ cells. Similarly, MPP6 a transcriptional marker of Tbet+ (TBX21) cells34 was downregulated in switched memory CD11c+, providing further evidence for the observed heterogeneity of CD11c+ cells in terms of expression of TBX21.32 Seven of the differentially expressed genes between the naïve-like CD11c+ cells and the switched memory CD11+ cells (CD27, LTB, TSC22D3, ACTG1, FCRL3, FCRL5 and KLF2) were also independently validated in the smaller dataset of Su et al. with significant adjusted p-values (Figure 5B) and many other important markers such as CXCR3 and MPP6 had the same direction of effects.
To our knowledge, this is the first study to be transcriptionally characterising these very rare subpopulations of cells after COVID-19 infection by leveraging information from two independent single cell technologies and using only publicly available datasets. Importantly, this subpopulation of CD11c+ cells has never been identified without prior sorting of B cells. These results have clearly demonstrated that the multimodal integration substantially improves our ability to resolve cell states, allowing us to identify and characterise previously unreported B cell subpopulations.
In the last decade, single cell methodologies have transformed all fields of biology. Mass cytometry and single cell RNA-Seq have been very widely used in the past, but the integration of the two modalities has not been widely explored so far. In this work, we show the unique advantages that can be gained from formally integrating these two modalities. We demonstrate that CyTOF datasets can be used to annotate and produce high quality imputed proteomics values for both matched and unmatched scRNA datasets. More importantly, we prove that the imputed values can be used to help define rare subpopulations in order to transcriptionally characterise them in depth using publicly available datasets.
The integration of modalities using imputed or projected values can give us unprecedented power to further our understanding of the transcriptional and proteomic immunological landscapes of health and disease and provide a wealth of interesting hypotheses to be tested, but caution is warranted when using this methodology without validation. We observed great potential in integrating well defined populations of cells, such as CD4 and CD8 T cells, but in cases where the common markers were not sufficient to separate the cellular heterogeneity, the agreement between the projected cellular annotations and the real ones were suboptimal (such as GDT cells; Figure 1B-D). Fundamentally, this integration will always depend on the quality and quantity of the common CyTOF-RNA features.
An additional limitation of this methodology is that the frequencies of rare subpopulations between different conditions cannot be estimated with high accuracy for medium size scRNA-Seq studies (Supplementary Figure 12, Extended data42). Populations that were less than 1% of the B cells had a high proportion of samples for which no cells could be salvaged for those subpopulations. However, we demonstrated that meta-analyses of RNA datasets can bypass this constraint and managed to unravel the transcriptional heterogeneity of these populations. Exploiting this methodology with the abundance of scRNA-Seq data from the Human Cell Atlas1 could provide endless opportunities to harness the differences between rare subpopulations in health and disease.
We should note here that the integration of CyTOF with scRNA-Seq datasets is not intended to substitute the invaluable information gained from multi-modal technologies such as CITE-Seq; measurements of antibody derived tags will inherently be more accurate than imputed values. However, there are instances where mass cytometry can provide imputed measures which would not be available in CITE-Seq, such as when rare subpopulations of cells are of interest, as we demonstrate in Figures 4,5. Additionally, intracellular protein modifications of cells, which are not possible to measure in CITE-Seq, could be projected to the RNA datasets, in a similar manner to our imputation of non-overlapping features CD45RA and CD45RO to identify epigenetic states of the cells. Furthermore, extending the integration of scRNA-Seq datasets with Imaging Mass Cytometry (IMC) datasets could facilitate new and exciting discoveries of spatial interactions of healthy and disease states.
In this article, we have successfully identified and characterised a previously elusive subpopulation of CD11c+ B cells. To our knowledge, this is the first study showing that imputed protein values from a CyTOF and scRNA-Seq integration can lead to an in-depth transcriptional characterisation of a heterogeneous rare subpopulation of cells. Even though the frequencies of these subpopulations vary between datasets, we show that the transcriptional signature that defines those cells can be robustly identified with this integration. To date, there are only a handful of scRNA-Seq experiments that try to describe this CD11c+ population but all are very limited in the numbers of samples and cells used and none of them has managed to isolate all three subpopulations of CD11c+ cells.17,35,40,41 In addition, in these studies the subpopulations of interest had to be sorted prior to sequencing to enrich for rare subtypes. Cell sorting, however, is a laborious process that can introduce bias in the process of selection of cells, especially in the case of less well-defined subpopulations, as is the case with CD11c+ subpopulations, explaining why none of these studies was able to identify all three subpopulations. Managing to couple phenotype-based studies with single cell transcriptomics in an unbiased way can give us unprecedented insights into the cellular machinery of all sections of biology.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)