Keywords
Single cell, multi-omics, spatial transcriptomics, FAIR, data analysis, data standards, training, computing infrastructure
This article is included in the Bioinformatics gateway.
This article is included in the ELIXIR gateway.
Single cell, multi-omics, spatial transcriptomics, FAIR, data analysis, data standards, training, computing infrastructure
Single-cell omics (SCO) is an umbrella term that encompasses multiple technologies that are able to profile various omic modalities at the single-cell level. These high-throughput single-cell approaches have rapidly become the method of choice over traditional bulk methods which average data across a population of cells. Single-cell approaches are better suited for characterizing many biological phenomena and exploring cellular heterogeneity such as characterisation of rare cell types and diverse cell states. Besides single-cell/nucleus RNA sequencing,1–4 SCO approaches include nuclear epigenetic profiling such as chromatin accessibility,5 histone profiling,6 DNA methylation,7 chromatin conformation8 as well as high throughput single-cell proteomics.9 Recent developments allow simultaneous profiling of two or more of the aforementioned modalities,10 opening up unprecedented opportunities to study diverse processes such as development, gene expression dynamics, tissue heterogeneity and disease pathogenesis. More recently, several approaches have been developed to deliver spatial resolution of single cell expression within tissues, adding another layer of complexity. While several grand challenges in exploratory data analysis remain,11 a parallel issue is the provision of infrastructure to support such analysis in the rapidly developing field.
New SCO profiling technologies are mushrooming, and new analysis methods are published weekly (Figure 1a). As the scale and modality of data sets grow, new computational methods are required. The data also need to be stored and annotated in a standardized manner in order to enable their reuse. This in turn adds an extra challenge and makes it hard for most institutes to handle alone, and calls for international collaboration on training, tools, compute, data, interoperability and standardization in SCO.
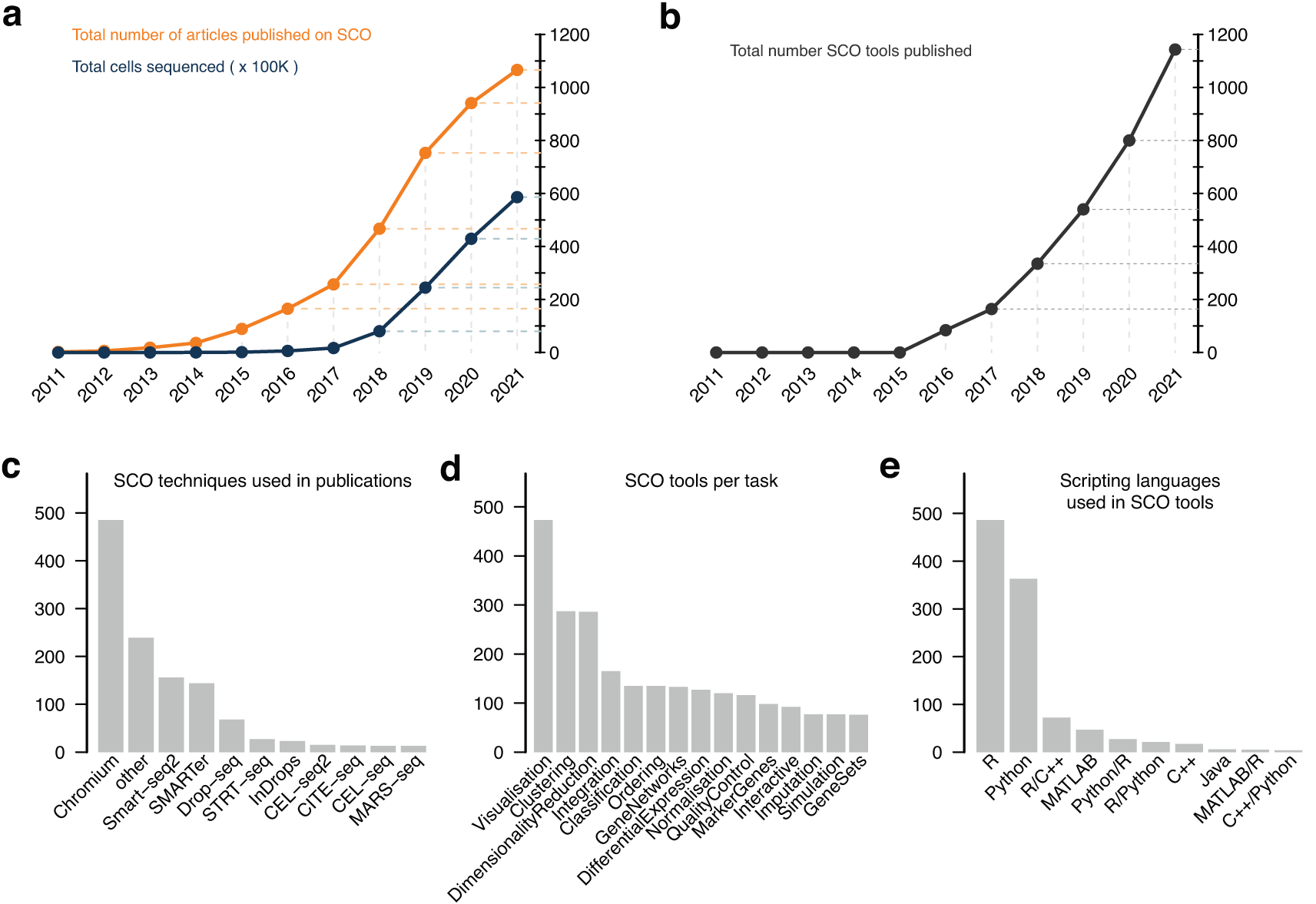
(a) Current count of articles using SCO technologies and cumulative number of cells sequenced and deposited in public databases. (b) Number of tools developed specifically to work with SCO. (c) Most common SCO molecular profiling technologies mentioned in publications. (d) Top 15 most targeted categories for software development in SCO. (e) Number of tools developed for SCO, split by which scripting languages are used. Data were taken from public databases.34,74 See the Data and Software Availability section for details.
ELIXIR, the European infrastructure for life sciences data, brings Europe’s national centers and core bioinformatics resources into a single, coordinated infrastructure.12 This intergovernmental organization currently has 23 Nodes, and facilitates collaboration between its member institutes and researchers with two intersecting organizational groupings: Platforms and Communities. ELIXIR Platforms (Data, Interoperability, Tools, Compute and Training) provide services, and ELIXIR Communities identify the needs of domain- or technology-specific research around a theme. There are currently 13 Communities ranging from Metabolomics and Proteomics to Federated Human Data and Galaxy.
Many ELIXIR Nodes already have single-cell facilities, and others are setting them up. The Nodes are facing a huge demand for single-cell data analysis and training, and some knowledge transfer between the Nodes already exists. Examples of past data analysis courses co-organized by ELIXIR Nodes are listed in Table 1. The Nodes have also co-organized workshops to discuss FAIR data management and best training practices for SCO. Together, 17 ELIXIR Nodes proposed to create the ELIXIR SCO Community to connect these grass-roots efforts and strengthen European and international cohesion in SCO.
Tutorial training style refers to teaching bioinformatics by following pre-made sequential analysis steps with code. PBL training style refers to teaching using project-based learning, where students develop their own analysis code to solve analysis tasks. All courses listed below were taught in English.
| Course name, link and year | ELIXIR node | Training style | Computing Language | Recorded lectures | FAIR Tool | Length | Technologies |
|---|---|---|---|---|---|---|---|
| Advanced topics in single cell omics (2021) | SE, CH | PBL | R and python | Yes | Docker | 5 days | scRNAseq, scATACseq, ST, Deep Learning |
| Single Cell School (2019) | SE, CH | Tutorial | R | No | - | scRNAseq, CyTOF, scProteomics | |
| Galaxy single-cell omics training materials (ongoing) | DE, UK, CH | Tutorial | - | Yes | Galaxy | 3-5 days | scRNAseq |
| Single-cell RNAseq analysis using R (2021) | UK EMBL-EBI | Tutorial | R | No | - | 5 days | scRNAseq |
| Gene Expression at Spatial Resolution (2021) | EMBL-EBI SE | Tutorial | - | Yes | - | 4 Days | Spatial transcriptomics (10X Visium) |
| Single-cell RNAseq data analysis with R (2019) | FI, SE, NL, DE, NO, FR | Tutorial | R | Yes | Conda | 3 days | scRNAseq |
| Single-cell RNAseq data analysis with Chipster (2020) | FI, LU | Tutorial | - | Yes | Chipster | 3 days | scRNAseq |
| Single-cell RNAseq data analysis with Chipster (ongoing) | FI, NL, SE, FR | Tutorial, eLearning | - | Yes | Chipster | 3 days | scRNAseq |
| Single cell RNA-seq analysis workshop (2022) | SE, NL | Tutorial | R and Python | Yes | Conda | 5 days | scRNAseq, ST |
Single-cell omics technologies have seen widespread adoption since its announcement as Nature Method of the Year in 2013.13 The most widely used SCO technologies are single-cell RNA-seq, single-nucleus RNA-seq, and single-cell ATAC-seq. The early days of these technologies were dominated by heterogeneous implementations of handling, preparation and sequencing protocols,14 which left its mark in the large number of software tools that had been developed, which in part led to lack of standardization of data and metadata. In recent years, we have seen a number of technology providers prevail for instrumentation (e.g. Fluidigm, 10x Genomics), reagents (e.g. ThermoFisher, QiaGen, Roche), and sequencing (e.g. Illumina, MGI, ONT and PacBio). This has led to the community converging around a few workflows based around the popular 10x Genomics Chromium and SmartSeq chemistries (Figure 1c), which have been exploited for large scale sequencing efforts such as the Human Cell Atlas (HCA)15 and Human BioMolecular Atlas (HuBMAP),16 that, in turn, have resulted in large investments into solving the sample handling, data integration and data management problems underpinning the vast array of data being generated.
While it has been a decade since the SCO technologies have taken the center stage in unraveling bio-molecular heterogeneity, technology developers are far from stagnant and we are seeing rapid evolution of these technologies. We witness the adoption of single-cell genomics (e.g. MissionBio Tapestri) and some progress in the field of single-cell proteomics.17 Single-cell multimodal omics was announced as Nature Method of the Year 2019.10 These assays provide multiple readouts that can be used to define cells. For example, antibody profiling (e.g. CITE-seq18) allows scientists to contextualize novel cell types and states in the context of well-established cell biology markers; immune repertoire profiling can also link how transcriptional profiles of immune cells differ based on the receptor specificity19; true multimodal omics such as simultaneous profiling of chromatin accessibility, DNA methylation and transcriptomics (e.g. scNMT-seq20) allows us to also decipher the regulatory changes that underpin the transcriptional landscape of cells.
In 2020, spatially resolved transcriptomics (SRT) was announced as Nature Method of the Year.21 These technologies fall under three broad areas of laser capture microscopy combined with single-cell sequencing (e.g. GeoMX DSP, Tomo-seq), in situ capture arrays (e.g. ST, Visium, Slide-seq, HDST), and image-based single-molecule expression quantifications (e.g. in situ sequencing,8 seqFISH+,22 Molecular Cartography,23 MERFISH24). Despite its youth, there are over 20 SRT profiling technologies,25 which are also expanding into other omics modalities, such as proteomics (e.g. CODEX26) and metabolomics.27 However, the community quickly realized the potential of this technology and early efforts were pushed by the Chan Zuckerberg Initiative to harmonize the field by funding efforts such as StarFish,28 a platform to uniformly process raw single molecule SRT data, and the SpaceTX consortium, which aim to benchmark and harmonize data from various SRT platforms and analytics methods. Efforts to make comprehensive cell atlases available to the community now facilitate more single-cell study designs to include perturbation and lineage tracing experiments (e.g. via CRISPR29–33).
While the standards for scRNA-seq have by-and-large converged, the extension of single-cell technologies to new modalities and experimental setups places even more emphasis on establishing adaptable and extensible standards.
There is a large number of single-cell analysis methods and tools available that cover a wide range of analysis steps. In January 2022, the scRNA-tools database recorded nearly 1,200 tools divided over more than 30 categories (Figure 1b and d).34 Computational and analytical challenges in single-cell genomics have been discussed extensively.11,35 The analysis steps vary depending on the modality of single-cell data. For the most widely used modality, transcriptomic data, the community has converged on a consensus regarding the analysis steps.36 Yet, even some foundational steps remain active areas for research, such as how to normalize scRNA-seq data37–39 or how best to perform differential expression analysis.40,41 Also, annotation of cell types varies drastically between studies, with many resorting to ad hoc decisions. A complete atlas of all cell types would be required to improve the standardization of cell-type nomenclature and ontologies (e.g. Cell Ontology, UBERON).42 Scientists analyzing SCO data need to have sufficient information on the strengths and limitations of the available analysis tools in order to select the most suitable ones for their data and purpose. However, systematic comparison of these tools is challenging, especially given the ever-increasing number of methods and their parameter combinations.
Studies have identified essential guidelines for benchmarking computational methods43–49 and reviewed published benchmarking studies of computational tools for omics data, highlighting the advantages and limitations of benchmarking across various domains of the life sciences.50 Systematic benchmarking frameworks can enable crowdsourcing and community challenges, which have been a successful means for fostering community creativity and expertise to address open problems.51 In a concerted effort to address “grand challenges” for the SCO community,11 the Open Problem in Single-Cell Analysis group52 is devising competitions to address those challenges, e.g. the Multimodal Single-Cell Data Integration competition (NeurIPS 2021).
Despite community efforts, the major challenges facing SCO benchmarking studies are the lack of appropriate experimental data and/or realistic simulated data that can be used for benchmarking, as well as the lack of agreed-upon measures to evaluate different methods. There is also a need for a common platform to conduct benchmark studies. The Open Problems NeurIPS challenge provides a leading example for evaluating methods using common datasets, performance metrics, as well as providing a compute infrastructure to run these methods. However, there is still a need for platforms that allow for continuous update of results as new tools and/or metrics become available and to dynamically respond to the needs of individual communities within the life sciences.
Currently available tools and pipelines differ in their usability. While the majority require programming knowledge, several pipelines provide GUIs for users without programming experience (e.g. Galaxy, Chipster).53,54 Most tools are available as R and Python packages or as a collection of scripts on GitHub (Figure 1e). To keep up with the technology developments, these methods and tools are continuously updated. Yet, maintaining tools and providing support is often challenging for research groups. Interoperability between methods and tools is limited despite efforts by popular packages such as Seurat55 to provide wrappers around other tools. However, frequent updates to tools to keep up with technology developments (e.g. updating single-cell objects to cater for multi-modal data) limits interoperability, emphasizing the importance of a concerted effort to address robust data and metadata standards.
The major factor in realizing interoperability is the definition and adoption of robust data format standards. While more than 1,000 SCO tools exist, there is broad acceptance of widely adopted raw data standards (e.g. FASTQ, FAST5, BAM, CRAM) and convergence to a few processed data formats (e.g. tab-separated files, AnnData, HDF5, loom, SingleCellExperiment, Seurat). The data formats and structures employed by some of the most popular tools for SCO data analysis55,56 have had to change to adapt to new technologies that rendered previous formats inadequate. While these changes in data formats are frustrating for maintaining data analysis workflows, they are necessary for keeping up to date with the rapid technological developments in this field. This places a strong emphasis on planning to adapt to changes by employing extensible structures that do not break the chain of backwards compatibility.
Furthermore, metadata standards and minimal reporting guidelines enable the appropriate archiving and subsequent reuse of SCO data. For some specific library construction or sequencing technologies, provision of platform specific metadata is routine and standardized (e.g. for the 10x Chromium), however additional care is required for in-house solutions and for reporting metadata for other parts of the experimental design. Establishing the Minimum INformation about a SEQuencing Experiment (MINSEQE) guideline was an important achievement for reporting metadata for sequencing data.57 Recently the Minimum Information about a Single-Cell Experiment (minSCe) guidelines were established,58 which defines 48 attributes that describe the biosource, isolation method, protocols, library construction, sequencing assay, raw data files and sequences, and cell- and sample-associated information derived from data analysis. However, as we move towards atlasing entire organisms and the rapid emergence of spatially resolved SCO technologies, further refinement of these minimal reporting standards is required to allow for describing common landmarks to facilitate integration of reference maps at differing scales into a single common framework, e.g. through the adoption of the Common Coordinate Framework59 that aims to uniquely and reproducibly define any location in the human body. A major consequence of establishing robust metadata standards is that they facilitate the establishment of SCO portals that provide access to uniformly processed data from a wide variety of SCO studies (e.g. the EBI Single Cell Expression Atlas, the Broad Single Cell Portal, and the HCA Data Portal). Such portals rely on accurate and sufficient metadata to enable appropriate processing of SCO data from a wide variety of studies in a uniform way.
Given the fast pace of technological developments in the SCO field, the community has identified that both adaptability and extensibility are key considerations in defining sustainable standards. This has been achieved in the field of medical imaging with the Digital Imaging and Communications in Medicine (DICOM) format,60 which has been constantly extended and updated without breaking backwards compatibility for nearly 30 years. However, this level of flexibility was only achieved by the third version of the DICOM standard, 10 years after its initial inception, and it was a concerted effort between medical and trade associations. Part of the successful adoption of the DICOM format is that despite all major medical imaging players having their own proprietary formats, they provide an interface to the DICOM format. In order for the SCO community to reach a similar level of interoperability as has been achieved in medical imaging, technology providers and tool developers should also either adopt the most common standards in the SCO community, or provide interfaces to them. While it is not clear how the current landscape of SCO data and metadata standards will stand the test of time, some aspects that will determine their success with be their ability to adapt to change (e.g. through using extensible formats such a JSON), used of controlled nomenclature (e.g. utilising ontologies for defining attributes), and adopting versioning (e.g. semantic versioning).
Upskilling life scientists to analyze SCO data is a moving target, given the fast development of the field. The cutting edge analysis methods for SCO data tend to be rather computationally complex, making them harder to grasp for life scientists who typically lack a solid background in mathematics, statistics and machine learning and often R/Python skills too.
Trainers, on the other hand, find themselves updating training materials constantly and, in general, struggle to keep up with the fast development of new analysis methods in order to choose what to teach. To make things worse, often only a small fraction of their working time is dedicated to training, or training is offered on a voluntary basis on top of their workload. It is therefore not surprising that even though single-cell courses are offered by several ELIXIR Nodes, many flavors of SCO are not yet covered. For example, courses on single-cell epigenetic, multi-omics as well as image-based spatially resolved SCOs are still rare. The demand for training continues to grow, but the lack of competent experts with enough training experience and time available is a major bottleneck in scaling up training provision. While pedagogical train-the-trainer (TtT) courses61,62 can empower experts to feel more comfortable to teach, the constant evolution of the SCO field can intimidate newcomers.
There are also more practical hurdles: the analysis of single-cell data requires a sophisticated computational environment with many tools and their dependencies, often requiring high-end computational resources. These environments have to be ready-to-go or at least easy to set up, and reproducible across heterogeneous hardware infrastructure, allowing the participants to re-run the practical and to analyze their (probably much larger) own data in their own setting. It is also challenging to find good training datasets that are small enough to be run in a class but meaningful enough to prove the concepts.
The ELIXIR SCO Community will bring together current efforts and produce guidelines and training. It creates a communication channel to exchange experiences, collect user requests and feedback and push for standards. Given its needs for training, tools, compute, data and interoperability, the SCO Community aligns well with all the ELIXIR Platforms. It also has synergies with the ELIXIR Human Data Communities and the Galaxy Community, as well as some ELIXIR Focus Groups like Cancer Data and FAIR Training.
Upskilling scientists in SCO data analysis and standards lies at the heart of the ELIXIR SCO Community, and particular efforts will be made to make the training scalable and FAIR in coordination with the ELIXIR Training platform (Table 2).
The SCO Community will ensure that training materials and expertise are shared efficiently and following FAIR and open research principles.63,64 We will collaborate with ELIXIR’s Training Portal TeSS65 to establish a well-curated SCO training portal, listing national and international training providers, web resources and upcoming training events. To help the current trainers and encourage new ones, we will annotate training materials with appropriate metadata, curate training datasets, provide detailed explanation on how to run courses, and share best practices and best ways to teach the more advanced concepts. In order to identify SCO areas which lack sufficient training, we will participate in designing the annual training gap survey by the Training Platform, and also perform more detailed SCO training surveys if needed. We will regularly host trainer workshops targeting the areas identified as lacking sufficient training to exchange experiences and discuss materials.
Anyone should be able to learn about SCO data analysis independently of time and place. To make the training scalable, lectures and video tutorials will be recorded for asynchronous learning, and combined into modular eLearning courses. Resources will be gathered and annotated on a single site for easy discovery. In addition to organizing training in SCO data analysis and standards to complement ELIXIR Nodes activities, we will provide training in best practices for trainers (TtT) to increase the number of expert trainers.
The course software installation challenge will be addressed together with the ELIXIR Tools Platform, as described below, using Conda environments, containers and Notebooks. Both Galaxy66 and Chipster54 offer specific training access and a comprehensive collection of training materials. There is no setup required, and the same environment is available when analyzing one’s own data after the course.
SCO data analysis typically requires a large number of tools and their dependencies. The installation challenge can be eased by providing Conda environments67 and containers68 for SCO, in alignment with the work developed in the Tools Platform’s Packaging, containerisation and deployment activity. Also, RStudio or Jupyter Lab based SCO Notebooks can be made to support courses and self-study. The Community will develop cloud-deployable analysis pipelines for SCO data and make them available also through the web-based Galaxy Single Cell Omics, Galaxy Human Cell Atlas project, and Chipster analysis platforms for researchers lacking programming skills. The analysis pipelines will be deposited in WorkflowHub69 for easy discovery, re-use and assessment.
The SCO Community will take several actions to address the aforementioned challenges in benchmarking. Liaising with data analysis experts, we will carefully curate data collections suitable for addressing specific tasks within the SCO data analysis workflow (e.g. multi-modal data integration, deconvolution of bulk data). For this, we will survey the landscape of existing benchmarking studies and identify the datasets they used and how they were evaluated. Whenever possible, our focus will be on real datasets rather than simulated ones, given the bias introduced by simulated data towards methods using the same underlying model. In order to address the lack of agreed-upon performance metrics to evaluate different types of methods, we will collect and curate existing metrics, and develop/suggest new measures when necessary (Table 2).
Regarding the need for a common platform to conduct benchmark studies, we will explore using OpenEBench.70 This ELIXIR benchmarking platform offers a flexible computational framework that allows individual communities to design and perform their benchmarking experiments. Communities are responsible for defining the reference datasets and the evaluation metrics and designing and developing evaluation workflows. Software developers are then able to use these workflows to evaluate their tools against the reference datasets, and the computed metrics are compiled, analyzed and publicly exposed in tables and visualizations. The results of the evaluation are then used by the community or any other OpenEBench user to decide which is the most suitable tool to do their analysis. The SCO Community will provide guidelines for the setup of single-cell benchmarking experiments. The guidelines have to cover three topics: 1) the scope of the benchmark, 2) the evaluation metrics that will be used to measure the performance of the tools and 3) the reference or gold standard datasets. The SCO Community will establish a benchmarking environment for SCO data analysis tools within the OpenEBench infrastructure, to facilitate a variety of community-driven challenges to address the diversity of the SCO applications.
The SCO community will perform periodic reviews of highly performant and rapidly adopted methods for registration in the bio.tools catalogue. To this end we will work closely also with the EDAM ontology to define single-cell specific keywords, which will help us not only to annotate the tools but also tag courses in TeSS.
The computing resource requirements of SCO data analysis increase constantly as the scale and modality of the data sets grow. The discussion between the SCO Community and the ELIXIR Compute Platform is therefore vital to ensure sufficient resources. The Community will also benefit from the Compute Platform’s Container Orchestration task, which will allow execution of containerised software tools and workflow workloads supporting public and sensitive data across ELIXIR Nodes. The ELIXIR Authentication and Authorisation Infrastructure (AAI) will be supported in the context of sensitive SCO data and whenever controlled access will be needed, we count on learning from the HCA’s experience on this matter.
The ELIXIR SCO Community will promote the development and usage of standards of metadata and file formats to ensure reproducibility of analyses and data reuse across biological and bioinformatics research communities. We will support existing efforts for aggregating and disseminating related metadata standards, e.g. from ArrayExpress, the HCA and further efforts. This is particularly important for emerging spatially resolved data, for which we aim to investigate efficient and scalable reporting structures, in line with current efforts in imaging and omics databases (Table 2).
An important consideration for sensitive human data is the General Data Protection Regulation (GDPR). To comply with the GDPR, raw human sequencing data deposited in EGA is protected and requires approval of the Data Access Committee Officer (DACO) as well as Data Transfer Agreements (DTA) outlining the conditions for allowing access to sensitive data. However, there is heterogeneous interpretation of the GDPR across Europe, and to facilitate this there have been a number of nationally Federated EGAs being established. Other non-human raw sequencing data would be deposited in ENA and would not be subject to these restrictions.
The ELIXIR SCO Community will encourage all data generators to ensure their data is available from an ELIXIR core data resource, or is deposited with a suitable ELIXIR core deposition resource, wherever possible, to ensure maximum data reuse and long term sustainability of all SCO data across the broader community. Via connections to key ELIXIR resources at EMBL-EBI (ENA, EGA, ArrayExpress and BioSamples database), we will promote discussions with these data resources to encourage the adoption and development of standards, where needed, to support the rapid pace of technology change in the single-cell field. We will leverage EMBL-EBI connections to the HCA Data Coordination Platform to broker the HCA data to ELIXIR core data resources and deposition resources.
The SCO Community will bring together data standardization efforts across Europe and combine them with global collaborations. The EMBL-EBI Node is a member of the global HCA community, whose mission is to create comprehensive reference maps of all human cells as a basis for both understanding human health and diagnosing, monitoring, and treating disease.15 It is also involved in the NIH-supported HuBMAP consortium,16 which develops tools to create an open, global atlas of the human body at the cellular level. The EMBL-EBI has already led an international effort to define the first guidelines for metadata standards of scRNA-seq experiments,58 involving members of the HCA and HuBMAP data platforms. As SCO techniques develop, we expect these guidelines to evolve to enable reproducible analysis of other methods, such as scATAC-seq, CITE-seq, single-cell HiC, to name a few.
Importantly, the ELIXIR SCO Community will align its activities with the LifeTime FET initiative,71 which combines single-cell multi-omics technologies with artificial intelligence and machine learning in order to revolutionize healthcare by tracking, understanding, and treating human cells during diseases. The LifeTime consortium includes over 90 research institutes and 70 supporting companies across Europe. Scientists from some of ELIXIR Nodes belong to both the LifeTime initiative and the SCO Community, thereby providing a direct link between them.
While HCA, HuBMAP and LifeTime focus on human cells, it is important to note that SCO technologies are used for different organisms, and thereby the ELIXIR SCO Community is not limited to human research. For example, the EMBL-EBI is also involved in the Fly Cell Atlas consortium.72
The training activities of the SCO Community will be enriched by collaboration with the Global Organization for Bioinformatics Learning, Education and Training (GOBLET).73 GOBLET’s mission is to cultivate the global bioinformatics trainer community, set standards and provide high-quality resources to support learning, education and training. The emerging SCO Community and GOBLET co-organized a global workshop for single-cell RNA-seq data analysis trainers in 2021. Sharing information about different training approaches, materials and datasets was considered very useful by the participants, and follow-up workshops are planned.
Finally, the SCO Community is discussing with the emerging SCO Community of Australian BioCommons, which is currently collecting user needs and finding solutions to the challenges identified, similar to us.
The SCO paradigm represents a revolution in the life sciences that pushes the boundaries of what can be explored, creating both new opportunities and challenges. We are witnessing increasing numbers of individual- and multi-omics modalities, and spatio-temporally resolved read outs. Both the rapid pace of advancement and adoption indicate that SCO will become the new normal in the life sciences. In the past five years, many ELIXIR Nodes have been working to assemble resources with the goal of developing future-proof guidelines and infrastructure as well as delivering training to SCO scientists. Here, we defined key goals at different infrastructural areas in order to create the ELIXIR SCO Community (Table 2) to ultimately strengthen current and foster new collaborations, and establish sustainable European and global frameworks for SCO research.
Data on scientific publications on SCO and number of cells sequenced was obtained from the Single-cell studies database.74 Data on SCO tools was taken from the publicly available repository of the scRNA-tools database.34
PC, PMP, IP, TB, JL, KH and EK conceptualized the study. PC performed data curation and visualization. KH performed project administration. PC, AM, RAC, PMP, LPS, AGU, LA, SCG, BS, MA, NI and EK wrote the original draft of the manuscript. PC, AM, PMP, LPS, AGU, CS, BS, PV, H-RH, BL, JMH, BG, WH, RK, TR, AB, PF, KH, NI, and EK reviewed and edited the manuscript.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)