Keywords
SARS-Cov-2, Family Cluster, Whole-Genome Sequence, Next Generation Sequencing, Breakthrough infection.
This article is included in the Nanopore Analysis gateway.
This article is included in the Genomics and Genetics gateway.
This article is included in the Coronavirus (COVID-19) collection.
SARS-Cov-2, Family Cluster, Whole-Genome Sequence, Next Generation Sequencing, Breakthrough infection.
SARS-CoV-2 was first detected in late 2019; however, its rapid mutations have resulted in several dominant strains that threaten the efficacy of current treatment and control protocols. D614G was the first reported mutation to rapidly emerge among the dominant viral strains worldwide.1 This mutation then increased and generated the emergence of the variants of concern, such as Alpha, Beta, Gamma, and Delta, which alternately became the dominant strain in many countries.1 These variants had been reported to have higher transmissibility, more severe clinical manifestations, and the ability to potentially escape the immunity conferred by the current vaccines.2
Lockdown and quarantine have been the main actions applied to contain the spread of SARS-CoV-2 in the population. Nonetheless, the household setting has been a critical SARS-CoV-2 transmission pathway.3,4 The rate of secondary transmission among household contacts has varied from 16.6% to 30%.3,5 Household transmission is more challenging to control, given the frequent unavoidable contacts among family members sharing the same home.6 In addition, transmission within households has been reported to be increased in families of greater size.7 The urban areas of Indonesia are less likely to have a family size of fewer than four individuals.8 The basic reproduction number in Jakarta during the early spread of COVID-19 was 1.75,9 which has made the household setting one of the vital arenas for COVID-19 transmission in Indonesia.
SARS-CoV-2 surveillance has been implemented in many referral laboratories in Indonesia since the middle of 2020, and it has supported researchers in examining the dynamics of SARS-CoV-2 variants throughout the year. This study was performed to assess the variation of SARS-CoV-2 strains and their clinical manifestations within family clusters in Jakarta, Indonesia.
This study was conducted after obtaining ethical clearance from the Research Ethics Committee of Universitas Indonesia (protocol number 20-05-0516). Patients were informed before providing written consent. All the patients involved in this study agreed to have their medical information and specimens used for analysis.
We selected naso-oropharyngeal swab specimens from family clusters positive for SARS-CoV-2 for whole-genome sequencing, choosing at least two patients to represent each family cluster. The specimens were stored in -800C from the date of detection to the sequencing process. The 200-μl of specimen extraction was performed according to the manufacturer’s instructions (PureLink™ Viral RNA/DNA Mini Kit, Thermo Fisher Scientific, USA). The real-time reverse transcription polymerase chain reaction (PCR) US Centers for Disease Control and Prevention validation test targeting N1 and N2 was performed according to the manufacturer’s instructions (SensiFAST™, Meridian Bioscience, USA).
The total RNA from positive extraction was then cleaned with DNase I and concentrated (RNA Clean & ConcentratorTM-5, Zymo Research, USA). The libraries were prepared according to the ARTIC multiplex PCR method, using an nCoV-19 V3 panel.10 The libraries were then processed with Oxford Nanopore’s GridION sequencer, employing MinKNOW version 20.06.9 and MinKNOW core version 4.0.11.10 High accuracy basecalling was conducted using Guppy version 4.0.11.11 All generated reads were assembled using EPI2ME Labs platform employing ARTIC workflow.10
The complete genome sequences were uploaded to GISAID for identification of clade, lineage, and mutations. The sequences were further annotated using SnpEff v4.3.12 The phylogenetic tree was built using Geneious Prime 2021.1.1, using UPGMA as the tree build method and Tamura-Nei as the genetic distance model, with NC_045512 as the reference sequence.
This study included 22 patients from eight family clusters, performed from January 2021 to August 2021. The patient characteristics are shown in Table 1. The phylogenetic tree is shown in Figure 1.
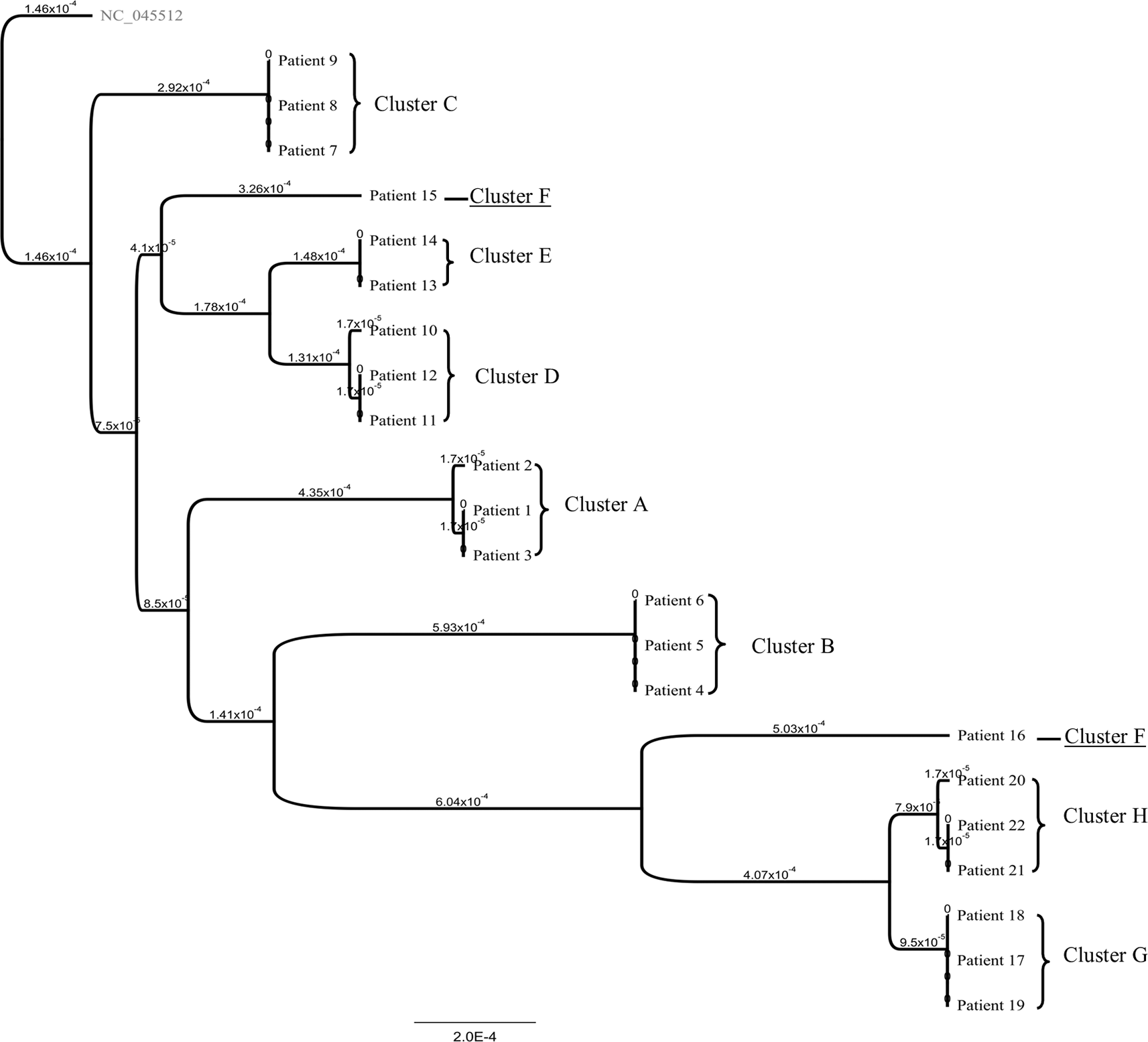
The source of infection in clusters A and D was unknown, and the infections were detected due to the screening program; the source of infection in clusters B, C, E, and F was a family member who had a history of working from the office during the pandemic; the source of infection in cluster G was a family member who had a history of traveling to another province via airplane for business assignments; and the source of infection in cluster H was the nanny who worked in the house during the day. Five patients had been vaccinated, with four of them completing the second dose.
In clusters C (4 of 4), D (5 of 5), E (5 of 5), F (3 of 3), G (4 of 4), and H (4 of 4), the whole family had become infected with SARS-CoV-2. For cluster A, only three of eight family members became infected with COVID-19. For cluster B, 4 of 5 family members caught COVID-19. The infection rate ranged from 37.5% to 100%.
Nineteen of the 22 patients isolated at home, with patients six, seven, 11, and 20 having pneumonia during their isolation, but who were not able to be admitted to the hospital because of the lack of available beds. However, these patients had access to the outpatient COVID-19 clinics to obtain doctor assessment, symptomatic medications, and radiology tests. The only patient with pneumonia who was able to be admitted to the hospital was patient three. Most patients did not have their blood tested during their early days after being detected positive due to difficulties reaching the healthcare service at the peak of the first and second wave. The Pango lineages circulating within the clusters were B.1.459, B.1., B.1.466.2, B.1.1.526, B.1.617.2 + AY23, and AY24. Most clusters had the same strain within each patient, except cluster F. For cluster F, patient 16 was detected positive after regular screening because he took care of patient 15, which explains the distance between the dates of the SARS-CoV-2 positive results between the patients.
This study had 2 dominant clades, GH and GK. The evaluation of the variation rates, transition-transversion (ti/tv) ratio, total single nucleotide polymorphism (SNP), total deletion, total variation, missense-silent ratio, spike (S) and nucleocapsid (N) gene nonsynonymous mutations, and total nonsynonymous mutation are shown in Table 2.
In addition, given that the spike gene had the highest number of mutations, the distribution of its nonsynonymous mutations within the patients is shown in Table 3.
| Patient | Cluster | A222V | D614G | D950N | E156G | F157del | G142D | H49Y | L18F | L452R | N439K | P26L | P681R | R158del | S689R | S494P | T19R | T478K | T716I | V1264L |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | A | + | + | + | + | |||||||||||||||
| Patient 2 | + | + | + | + | ||||||||||||||||
| Patient 3* | + | + | + | + | ||||||||||||||||
| Patient 4 | B | + | + | + | + | |||||||||||||||
| Patient 5 | + | + | + | + | ||||||||||||||||
| Patient 6* | + | + | + | + | ||||||||||||||||
| Patient 7* | C | + | + | |||||||||||||||||
| Patient 8 | + | + | ||||||||||||||||||
| Patient 9 | + | + | ||||||||||||||||||
| Patient 10 | D | + | + | + | ||||||||||||||||
| Patient 11* | + | + | + | |||||||||||||||||
| Patient 12 | + | + | + | |||||||||||||||||
| Patient 13 | E | + | + | + | ||||||||||||||||
| Patient 14 | + | + | + | |||||||||||||||||
| Patient 15 | F | + | + | + | ||||||||||||||||
| Patient 16 | + | + | + | + | + | + | + | + | + | + | + | |||||||||
| Patient 17 | G | + | + | + | + | + | + | + | + | + | + | + | ||||||||
| Patient 18 | + | + | + | + | + | + | + | + | + | + | + | |||||||||
| Patient 19 | + | + | + | + | + | + | + | + | + | + | + | |||||||||
| Patient 20* | H | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| Patient 21 | + | + | + | + | + | + | + | + | + | + | + | + | ||||||||
| Patient 22 | + | + | + | + | + | + | + | + | + | + | + | + |
Four clusters in our study had an index patient with a history of working in an office. Cases of SARS-CoV-2 infection from office workers outside the healthcare setting were scarcely reported.13 Six of eight family clusters had a 100% infection rate, showing the efficacy of SARS-CoV-2 transmission within the family group. As reported by several studies, the household infection rate ranged between 75% and 100%.3,14 The high infection rate within households combined with poor preventive measures in the workplace could generate a spike in COVID-19 cases.
This study also found SARS-CoV-2 infections in patients who had been vaccinated (breakthrough cases). Infection after complete vaccination was possible, no matter which type of vaccine had been administered.15,16 Duarte et al. reported the breakthrough cases mainly were mild, and that other factors might have influenced the symptomatic infections, given that they were not correlated with lack of vaccine-induced immunity.15 Although our study could not represent the general population, the concern of vaccine efficacy in the face of new variants with more mutations has become relevant since Christensen et al. reported an increase in breakthrough cases caused by the Delta variant (as part of the GK clade).17 Therefore, preventive measures such as wearing a mask are still necessary, even after full vaccination.
The clinical manifestation varied within the patients in each family cluster. This situation was in concordance with several case reports, which have mentioned variations in disease severity within a family cluster.18 However, seven clusters were monophyletic, which showed that the same virus could generate a different outcome. Furthermore, we found the virus genome of the patients with pneumonia was similar to the other patients without pneumonia (Table 1, Figure 1). No specific variation or combination in the genome mutation could generate a particular clinical outcome (Table 3). The D614G spike mutation was also distributed evenly within the patients. This finding was in concordance with Korber et al. and Lorenzo-Redondo et al., who noted that the D614G mutations did not correlate with more severe outcomes.19,20
Our study showed a ti/tv ratio decline between the GH and GK clades. This decline was in concordance with the study by Duchêne et al., which revealed the ti/tv scaled negatively over time, especially for rapidly evolving RNA viruses.21 We also found an increase in the GK total spike gene mutations compared with the GH clade. Given that the GK clade appeared later in our study, it was expected to have more mutations because the RNA virus would utilize various mechanisms of genetic variation to guarantee its survival.22 The evolving behavior can facilitate the virus to evade the current vaccine-generated immunity, resulting in breakthrough infections.
One limitation of our study was the small number of patients and family clusters. This study could not provide serological test results, especially from patients with vaccination history, given that there were no guidelines to confirm an immune response after vaccination. Our study showed that the same virus within a cluster could generate various clinical outcomes. Our observation showed the SARS-CoV-2 transmission in the household setting through the workplace, which could be a common pathway in the future of COVID-19 once the pandemic status is lifted. Although vaccination is expected to reduce the burden of COVID-19, control measures are crucial, given that breakthrough infections due to a new variant are evident.
The virus genome included in this study was published at GISAID with accession ID:
EPI_ISL_2692986, EPI_ISL_2692987, EPI_ISL_2692989, EPI_ISL_2993633, EPI_ISL_2993659, EPI_ISL_2993632, EPI_ISL_2692990, EPI_ISL_2692991, EPI_ISL_2692992, EPI_ISL_2993434, EPI_ISL_2993539, EPI_ISL_2993658, EPI_ISL_2692909, EPI_ISL_2692981, EPI_ISL_2692983, EPI_ISL_3000155, EPI_ISL_3456060, EPI_ISL_4847411, EPI_ISL_4847413, EPI_ISL_5054899, EPI_ISL_5054915, EPI_ISL_5054918.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)