Keywords
Saccharomyces cerevisiae INVSc1 protein expression, EGFP protein, protein transductions.
This article is included in the Cell & Molecular Biology gateway.
Saccharomyces cerevisiae INVSc1 protein expression, EGFP protein, protein transductions.
Enhanced green fluorescent protein (EGFP) has often been used as a reporter for protein transport, especially in eukaryotic cells such as mammalian and yeast cells. EGFP was derived from green fluorescent protein (GFP), a commonly used reporter protein that does not require a substrate for it to fluoresce. The EGFP gene is a mutated version of the GFP gene, with P64L and S65T mutations. These mutations cause a higher emission intensity in EGFP relative to that of the wild-type GFP, and better thermal stability of the EGFP protein, especially in eukaryotic cells.1 EGFP can be used as a marker in optimizing protein isolation procedures and their functions. The fluorescence makes it easier for us to observe the localization of the expression of the protein. The intensity of EGFP fluorescence can also indicate the level of expression of the protein in a system. EGFP can also be used to study protein transfer, such as analysis of the work of a protein transduction system for cell reprogramming purposes.
One of the uses of cell reprogramming is manufacturing induced pluripotent stem cells. Induced pluripotent stem cells are expected to be used as therapeutic cells for patients. Cell reprogramming can be done using protein transduction, viral transduction, or chemicals. Cell reprogramming by viral transduction can activate oncogenic genes, whereas chemical methods are highly unspecific and cannot be interpreted clearly.2,3 On the other hand, protein transduction is reputed as a harmless method to deliver protein precisely. However, protein transduction has its disadvantages, such as low transduction efficiency in host cells. Therefore, research is needed to increase transduction proteins by modifying the target protein.
Polyarginines are short polypeptides classified as non-amphipathic cell-penetrating peptides (NaCPP) and have cationic characteristics, including binding anionic lipid membranes; therefore, polyarginine can enter through the plasma membrane of eukaryotic cells. As a result, the NaCPP class is less toxic than the primary amphipathic and secondary cell-penetrating peptide (CPP). The NaCPP class uses endocytosis mechanisms with low concentrations for cellular uptake.4 The amount of added arginine amino acid residues affect the cellular uptake's efficiency level, whereas more arginine residues have a better efficiency level. Adding more than 15 amino acid residues decreases intracellular delivery efficiency.5 Proteins fused to polyarginine are more efficiently transduced into human iPS cells compared to those without the addition of CPP.6
In this research, we designed and constructed two proteins, EGFP and EGFP-PolyR. EGFP-PolyR is an EGFP protein with the addition of eleven arginine residues (polyarginine – polyR) at the C terminal of EGFP to be used as a transduction protein model for cell reprogramming purposes. The gene encoding EGFP and EGFP-PolyR was synthetically constructed and expressed in Saccharomyces cerevisiae INVSc1. The study aimed to investigate the pattern of EGFP-PolyR protein expression in S. cerevisiae INVSc1 compared to EGFP. This paper analyzes and discusses some of the possible improvements of EGFP and EGFP-PolyR protein expression in S. cerevisiae INVSc1.
The gene encoding EGFP (GenBank Accession #U55762) and EGFP- PolyR (was synthetically made by IDT Custom Gene Synthesis cloned in a plasmid called pEGFP- PolyR, in such a way that it allowed sub-cloning of the gene with and without the polyR sequence, by using a different combination of restriction enzymes. In addition to the 11× arginine residues (polyR), codons encoding 6× histidine residues (6×His-tag) were also added downstream to the polyR codons. The synthetic gene was sub-cloned into pYES2CT, a Saccharomyces cerevisiae expression plasmid, during which the polyarginine codon either remained fused to the EGFP gene or was removed, creating recombinant pYES2CT pYES2CT-EGFP-PolyR or pYES2CT- EGFP, respectively. Since the pYES2CT plasmid has its own 6×His-tag downstream to its Multiple Cloning Sites (MCS), positioned in-frame with the EGFP and EGFP-PolyR codons, the expressed EGFP was expected to have one 6×His-tag, whereas the EGFP-PolyR were expected to have two.
The intended digested fragments sized 821 bp (EGFP-PolyR) and 733 bp (EGFP), were gel-purified using GenepHlow Gel/PCR kit (DFH004) (GeneAid, Taiwan) with 20 μL elution buffer. The DNA concentration was measured using a BioDrop-Duo UV/Vis spectrophotometer (Thomas Scientific, United States). Then, the DNA was stored at -20°C. Ligation was carried out following the T4 DNA ligase procedure as described by the manufacturer (Promega, United States). The ligation was performed at 4°C overnight, with a plasmid-insert volume ratio of 1:10.
The transformation into Escherichia coli TOP10 was carried out following the method of Abyadeh et al. (2017).7 Recombinant E. coli cells were cultured in LB with 100 μg/mL ampicillin. Transformation by heat shock was carried out using the Scilogex SCI-100HCM-Pro Digital Thermal Mixer (United States) at 42°C for 90 seconds. The selection of transformants was performed on LB agar with100 mg/mL ampicillin (LB Agar+Amp). Colony PCR was performed on selected transformants with pYES2CT-specific primers (Forward: 5’-AATATACCTCTATACTTTAACGTC-3'; Reverse 5’-GAGGGCGTGAATGTAAGCGTGAC-3') to verify the presence of intended inserts. PCR was done using Biometra Tone Thermal Cyclers (Analytik Jena AG, Germany) and carried out following the procedure from MyTaq Red Mix (Bioline, UK) manufacturer, for 35 cycles, with predenaturation (95°C) for 1 minute, denaturation for 15 seconds, annealing (55°C) for 15 seconds, elongation (72°C) for 10 seconds, and post elongation for 5 minutes.
pYES2CT-EGFP- PolyR and pYES2CT-EGFP plasmids were isolated using' WizardPlus SV minipreps DNA purification kit (A1330) (Promega, United States)' after being propagated in E. coli TOP10. Isolated plasmids were used to transform Saccharomyces cerevisiae INVSc1 with the lithium acetate (LiAc) method.8 Transformants were selected on minimal medium (MM) agar without uracil. Plasmids were isolated from transformant colonies using the MP Biomedicals yeast plasmid isolation kit (MP Biomedicals, United States, Cat. 112069400). PCR was carried out to confirm the success of the transformation using Biometra Tone Thermal Cyclers (Analytik Jena AG, Germany) and carried out following the procedure from MyTaq Red Mix (Bioline, UK) manufacturer, for 35 cycles, with an annealing temperature of 55°C. Colony PCR was performed on selected transformants with pYES2CT-specific primers (Forward: 5’-AATATACCTCTATACTTTAACGTC-3'; Reverse 5’-GAGGGCGTGAATGTAAGCGTGAC-3'), to verify the presence of intended inserts. PCR was done using Biometra Tone Thermal Cyclers (Analytik Jena AG, Germany) and carried out following the procedure from MyTaq Red Mix (Bioline, UK) manufacturer, for 35 cycles, with predenaturation (95°C) for 1 minute, denaturation for 15 seconds, annealing (55°C) for 15 seconds, elongation (720C) for 10 seconds, and post elongation for 5 minutes.
S. cerevisiae's fast-growing diploid strain INVSc1 (Invitrogen Corporation) was used as a host cell for protein expression. The yeasts were cultured in yeast-extract peptone dextrose (YEPD) medium. Recombinant cells of S. cerevisiae INVSc1 were cultured in a minimal medium (MM) without uracil. Protein expression followed the user guide of the pYES2CT plasmid (Invitrogen, 2003), with several modifications. A single colony of recombinant S. cerevisiae INVSc1 carrying pYES2CT-EGFP or pYES2CT-EGFP- PolyR was inoculated into 3 mL of MM medium; the culture was then incubated at 30°C 230 rpm for 48 hours. After that, a 1-mL culture was taken to measure its OD600 value. Culture with OD600 = 0.4 were subcultured into 100 mL of induction medium (MM medium containing 2% galactose and 1% raffinose). After adding the induction medium, the cultures were further incubated for 48 to 72 hours. The culture was centrifuged at 5000 rpm for 10 minutes to harvest and stored in a freezer at -80 0C until further use.
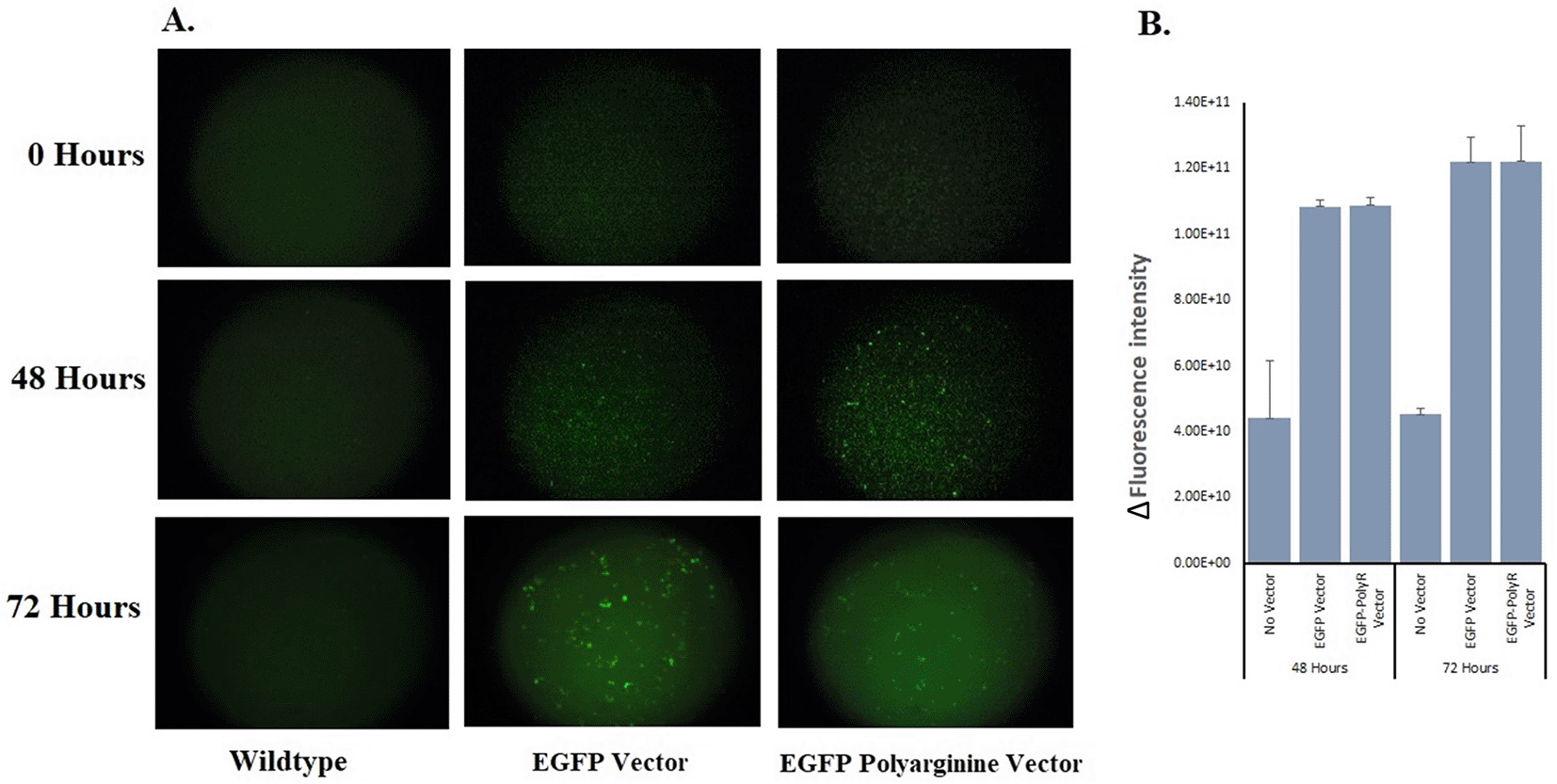
A 1 mL 10× diluted culture was centrifuged at 5000 rpm for five minutes. The pellet was then fixed with 500 μL 3.7% formaldehyde for 30 minutes. The cell suspension was centrifuged at 5000 rpm for 5 minutes, and the supernatant was discarded. as the remaining pellet was resuspended in 1 mL PBS 1×. A 50-μL fixed-cell suspension was placed in a well of 96-well plates (Thermo Scientific) to visualize the EGFP fluorescence under an inverted fluorescent microscope (Zeiss Axiovert 40 CFL, UK), (40× magnification, λex 488 nm and λem 507 nm). Fluorescent intensity was measured using ImageJ software, and delta intensity was calculated as follows:
where,Intensityt is the fluorescent intensity at time t post-inoculation to induction medium
Intensity0 is the fluorescent intensity at time 0 post-inoculation to induction medium
Lysis was carried out by sonication in combination with incubation in lyticase (Sigma-Aldrich). Pellet from 50 mL culture was prepared in 50 mL tubes, then 10 mL breaking buffer (1 mM EDTA, 5% glycerol, 1 mM PMSF, and 50 mM sodium phosphate, pH 7.4) was added, and the mixture was homogenized. Lysis was performed by sonication at 10 kHz for 10 minutes, alternating every 15 seconds between sonication and cooling on ice. When lyticase was used, sonication was performed after incubation in lyticase. After sonication, the mixture was centrifuged at 10,000 rpm for 10 minutes. Protein concentration was measured using the Pierce BCA Protein Assay kit (ThermoScientific, United States). The protein was concentrated by centrifugal microfiltration (Merck Amicon Pro Purification System, filter Molecular Weight Cut-Off (MWCO) 3 and 5 kDa).
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out following the modified Iqbal and Ahmad (2016) method. The protein sample was mixed with 6× SDS gel loading buffer (600 mM Tris-Cl pH 6.8, 12% SDS, 30% glycerol (v/v), 0.03% bromophenol blue, and 20 mM DTT) and run on 10 % SDS-PAGE gel at 100 V for 110 minutes until the bromophenol blue dye reached approximately 0.5 cm from the lower limit of the gel.9
Western blotting was also carried out following the method of Iqbal and Ahmad (2016), with several modifications. Protein was transferred from gel to membrane (transfer buffer: 300 mM glycine, 300 mM Tris, 0.05% SDS) using Pierce Power Blotter (Thermo Scientific, United States) set at 25 kV, 1.3 A for 25 minutes. Then, the membrane was washed six times with PBST (PBS, 1% Tween-20) before applying the primary antibody (Anti-6×His-Tag monoclonal antibody (Invitrogen, catalog number MA1-21315) in blocking buffer (5% skim milk (Merck, Germany) in PBST), with 1:3000 ratio). Then the membrane was incubated at 4°C overnight before applying the secondary antibody (Goat anti-mouse IgG HRP, Invitrogen, catalog number 31430 in blocking buffer, with a ratio of 1:20,000 ratio) for 60 minutes with shaking (70 rpm). Then, the substrate (SuperSignal West Femto Luminol, Thermo Scientific, United States) was used. The duration of the exposure was 20 minutes.
Two hundred microliter solution to be measured was placed in a well of the 96-well fluorescent plate. Measurement was performed using a fluorimeter set at λex 488 nm and λem 507 nm.
Amino acid composition and physicochemical properties were analyzed using ProtParam,10 and hydrophilic amino acids were analyzed using ProtScale.10 The isoelectric point was measured using the protein Isoelectric Point Calculator.11 The tertiary structure was modeled via i-Tasser12 and AlphaFold,13,14 and the Protean3D was used for viewing to analyze whether the histidine and polyarginine were located in the outer regions.
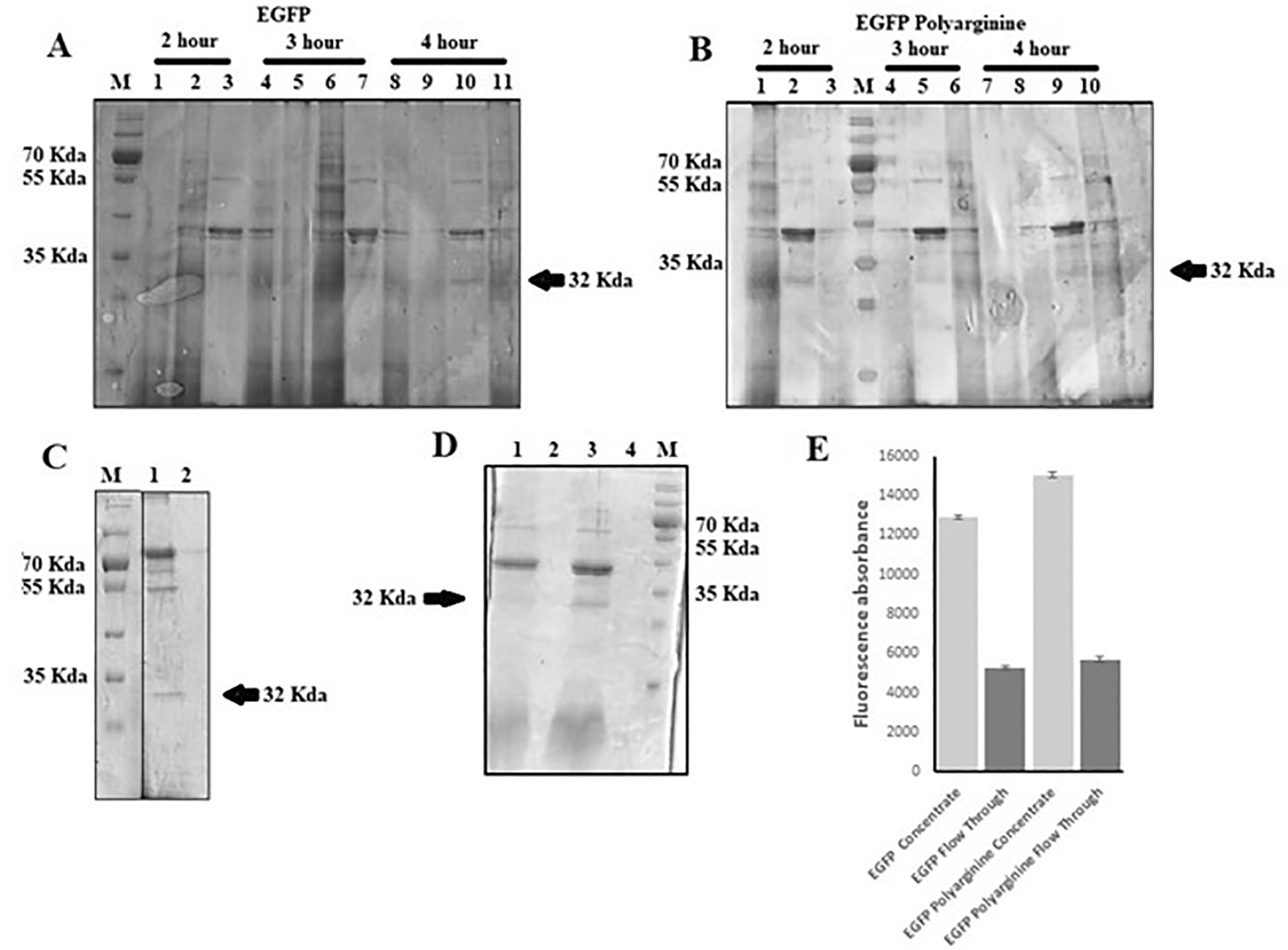
After digests and ligation processes were successfully performed (Figure 1 and Figure 2A), the ligation products, hereby called pYES2CT-EGFP and pYES2CT-EGFP-PolyR, for plasmid carrying egfp and egfp-polyR respectively, were propagated in E. coli TOP10. Plasmids isolated from this E.coli TOP10 that had been confirmed to have the correct size (Figure 2B) and carried the genes (Figure 2C) were then used to transform S. cerevisiae INVSc1. Screening of S. cerevisiae INVSc1 transformants was performed by PCR of purified plasmids. PCR results of positive colonies are presented in Figures 2D and 3.
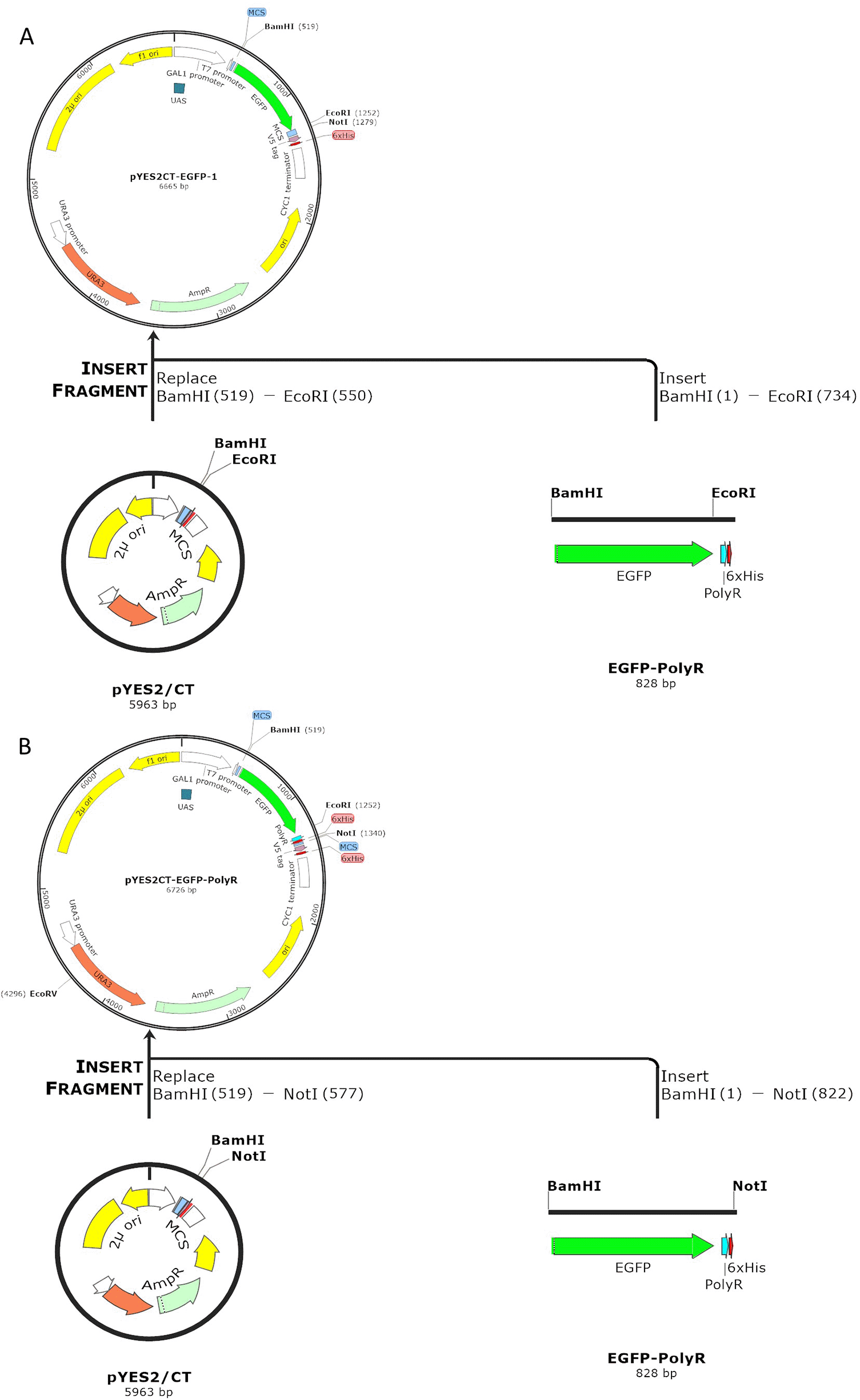
Insertion of EGFP-PolyR gene was performed using the enzymes BamH1 (position 519) and Not1 (position 577) (A), whereas EGFP gene was inserted into pYES2CT plasmid using BamHI and EcoRI, thus leaving the PolyR fragment out (B)
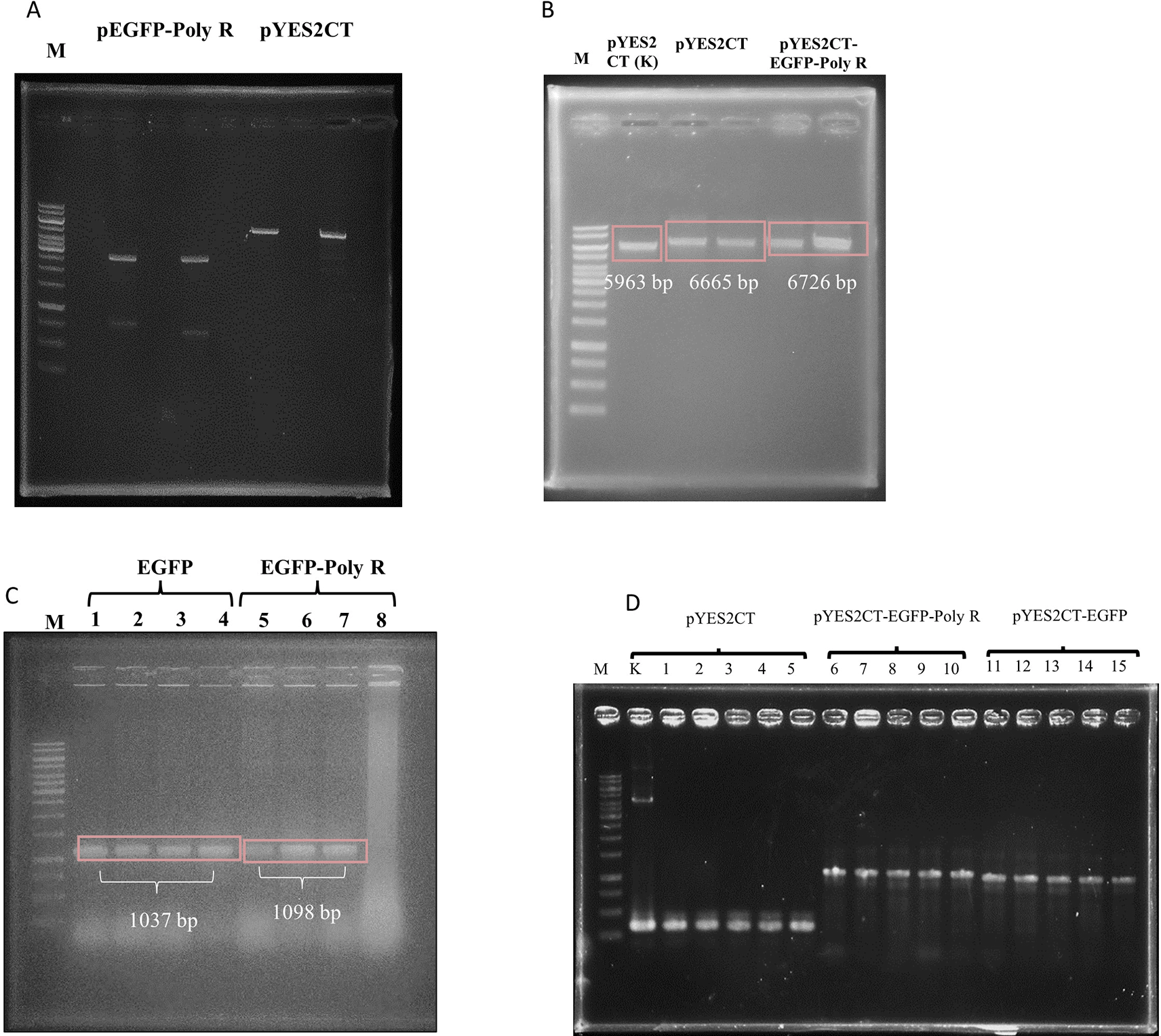
pEGFP-Poly R (lane 5) and pYES2CT (lane 7) were cut with restriction enzymes EcoRI and BamHI, where pEGFP-Poly R produced egfp gene (733 bp band), and pYES2CT produced vector (5905 bp band) (A). The 822 and 733 bp DNA were isolated from gel, and ligated to the 5932 and 5905 bp bands, respectively. Single digest (BamHI) of plasmids isolated from E.coli TOP10 transformed with pYES2CT-EGFP (lane 3 and 4) and pYES2CT-EGFP-Poly R (lane 5 and 6), in comparison to wild type pYES2CT (lane 2), showed that the plasmid sizes were correct (B). Also, colony PCR of E.coli confirmed the presence of the intended genes as 1037bp and 1098bp bands, representing the egfp and egfp-polyR, respectively (C). Transformed yeast colony screening was performed by PCR of plasmids purified from the yeast, using pYES2CT-specific primers. As a control, pYES2CT plasmid was used (lane K). Lanes 1-15 showed PCR amplification results of plasmids isolated from S. cerevisiae culture, transformed with pYES2CT (lanes 1-5), pYES2CT-EGFP-Poly R (lanes 6-10), and pYES2CT-EGFP (lanes 11-15). Amplification of pYES2CT without insert (lane 1-5) shows the same size amplicons as a positive control (K), which are 334 bp, amplification of pYES2CT-EGFP-Poly R produced amplicons of 1097 bp (lane 6-10), while the pYES2CT-EGFP produced amplicons of 1036 bp (lane 11-15) (D). Lane M is GeneRuler 1kb DNA Ladder (Thermo Scientific SM0312).
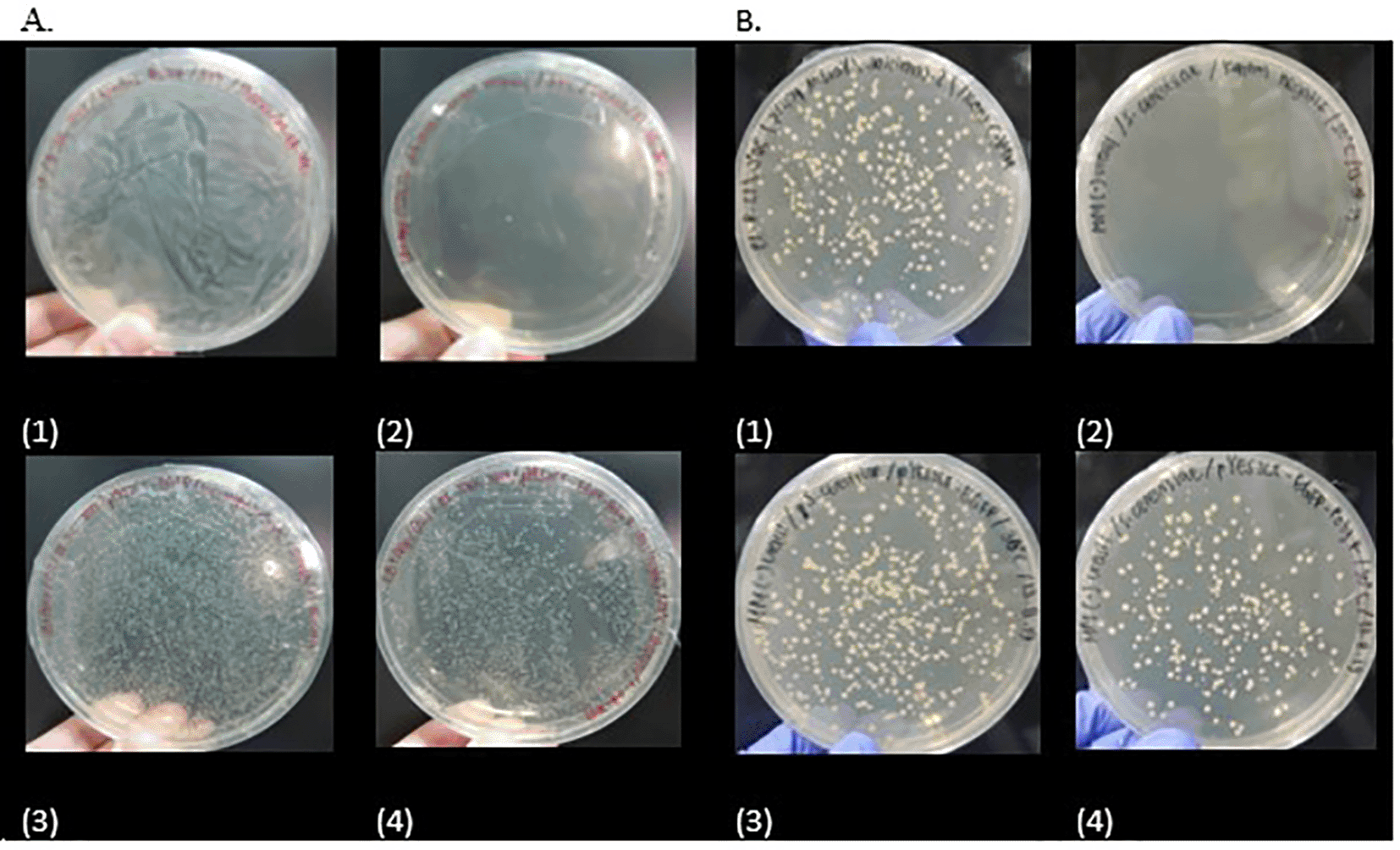
Transformation positive controls are shown in A1 and B1, whereas the negative controls are in A2 and B2.
The growth curve showed that post-induction S. cerevisiae INVSc1 stayed longer at the exponential phase when not carrying any plasmid in comparison to the same strain with pYES2CT plasmids (Figure 4). The plasmid-carrying yeasts seemed to reach stationary phase 24 hours after inoculation to induction medium (Figure 4B and C), whereas the wild-type culture stayed at exponential phase for up to 64 hours (Figure 4A) before slowing down its growth. No significant growth of the recombinant yeasts was observed between 48-72 hours post-inoculation to the induction medium (Figure 4D).
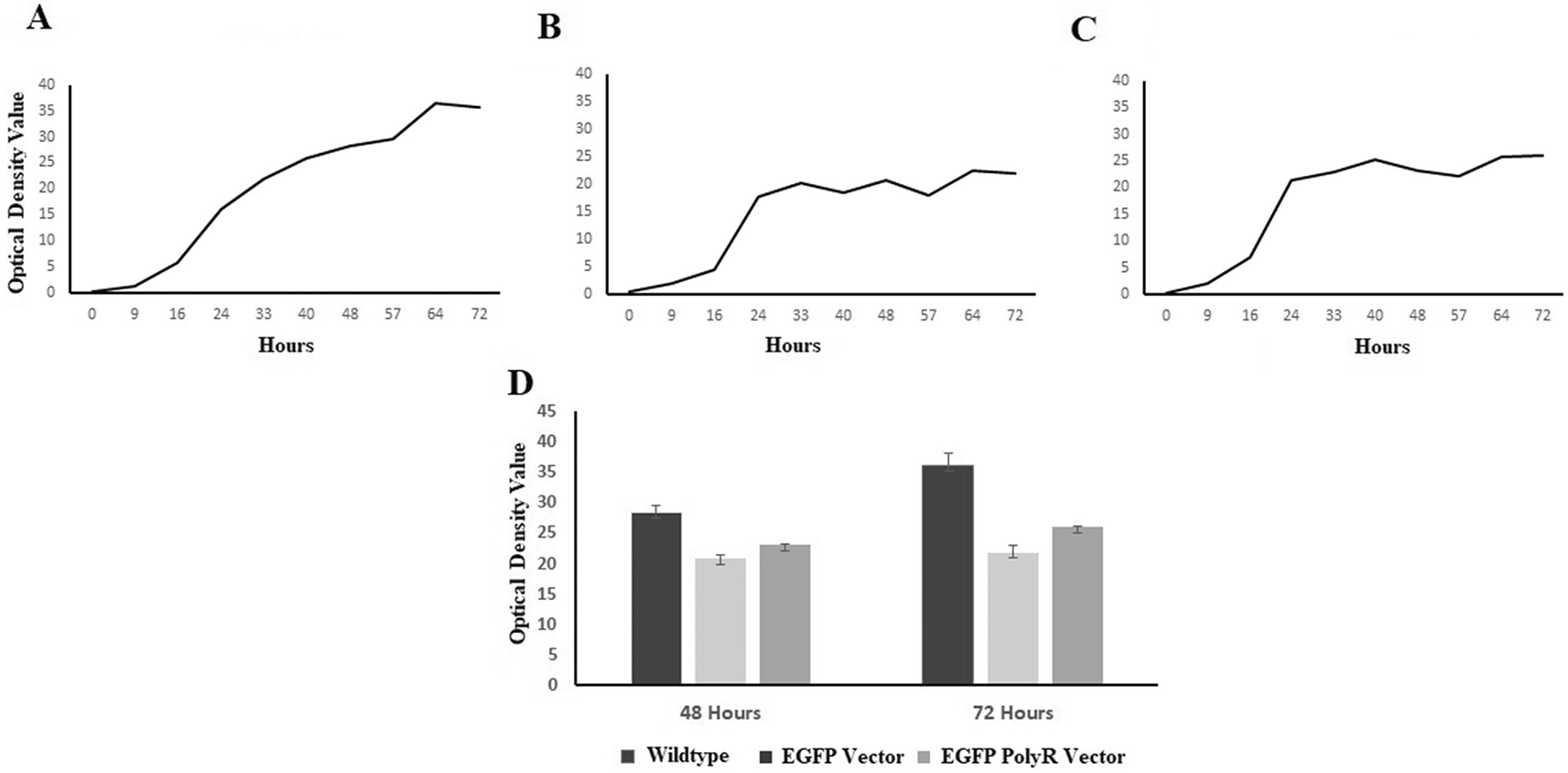
Optical densities were measured at λ=660nm, 48 hours, and 72 hours post-inoculation to induction medium of the wild-type and recombinant S. cerevisiae (D).
The fluorescence produced post-inoculation to induction medium by each recombinant yeast and wild-type yeast is presented in Figure 5A. The Δintensity was calculated and presented as a graph in Figure 5B. No significant fluorescence increase was seenwas observed between 48 – 72 hours in either the recombinant yeast carrying pYES2CT-EGFP or pYES2CT-EGFP-PolyR plasmid. There was no significant difference in intensity expressed by different constructs either.
Western blotting using anti-his-tag mouse monoclonal antibody (primary antibody) and Goat anti-mouse IgG HRP (secondary antibody) proved that the EGFP-PolyR and EGFP proteins were expressed in S. cerevisiae INVSc1 at similar but relatively low levels (Figure 6).
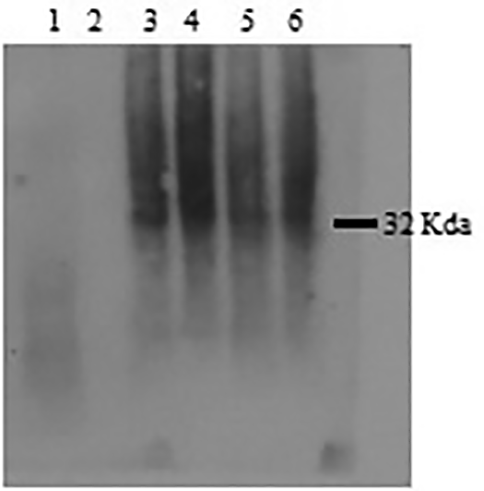
The Western blot was performed using an anti-his-tag antibody as the primary antibody.
A combination of lyticase treatment and sonication, as well as using sonication only, was attempted to extract the expressed proteins. We also attempted variations of incubation length (two, three, and four hours) in lyticase combined with five minutes of sonication. The results showed that, although the addition of lyticase increased the protein yield, in comparison to using sonication only (as observable from Figures 6A laanes 8 and 10), prolonging the incubation time in lyticase had no effect on protein extraction (Figures 7A and 7B). Attempts to concentrate protein by microfiltration using a filter with MWCO 3 and 5 kDa did not show a significant difference (Figures 7C and 7D). Measurement of fluorescence using a fluorimeter showed that concentrating the protein using a 3 kDa filter managed to retain most of the protein in the retentate (Figure 7E).
The properties of the proteins were analyzed using ProtParam and Isoelectric Point Calculator, and the results are presented in Table 1.
AA: amino acids; pI: Isoelectric point.
Modeling of the proteins was performed using iTasser, and the result is presented in Figure 8. The models show that in both proteins, EGFP and EGFP-PolyR, the 6xHis-tag is located outside the protein nucleus. Structural alignment using Protean3D software predicted that the presence of PolyR indeed affected the conformation of the fused protein, although not within the EGFP domain itself (Figure 7E and F).
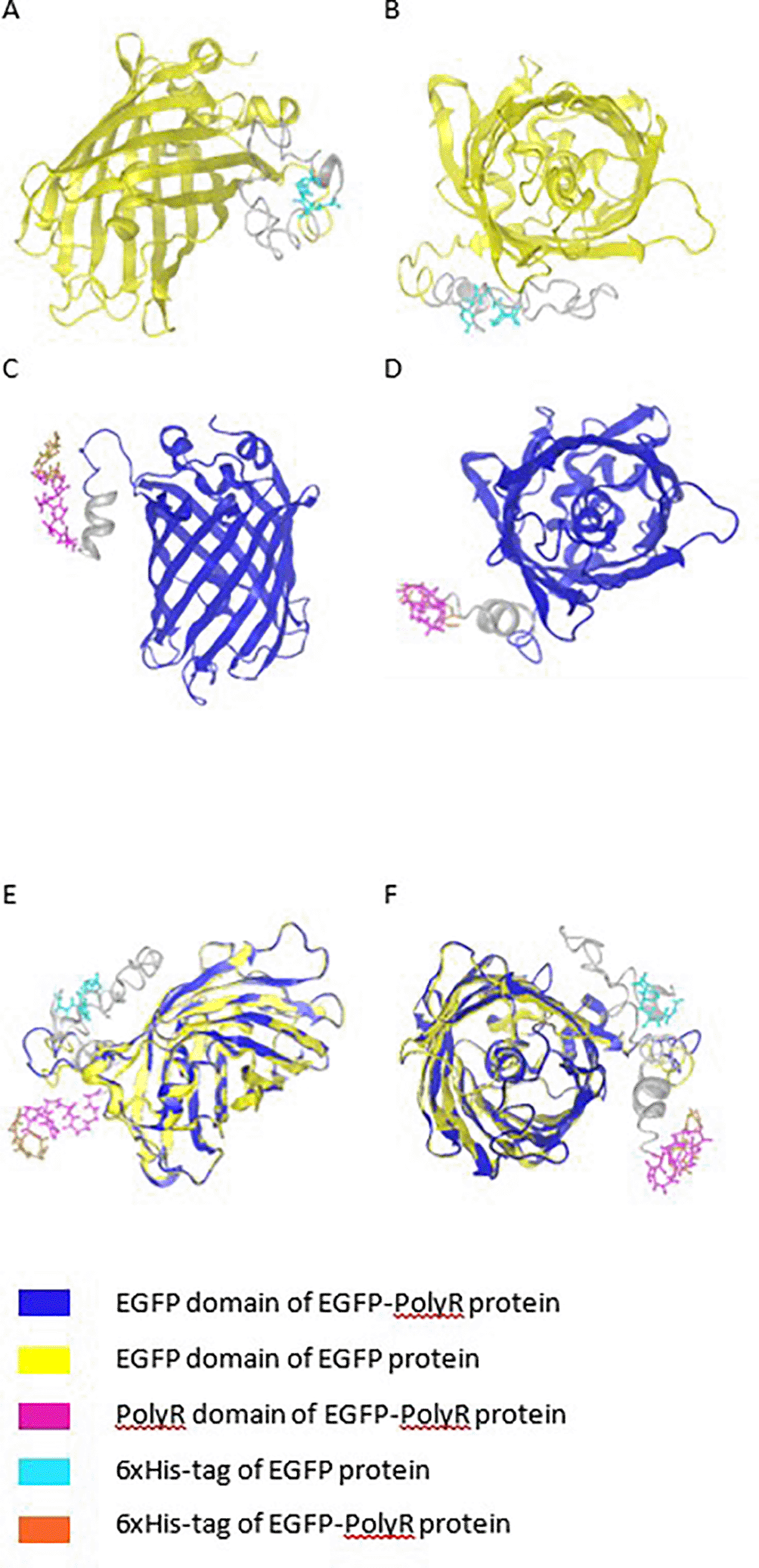
Alignment results of the proteins tertiary structures (E and F).
In this study, PolyR was added to EGFP protein to function as a cell-penetrating peptide (CPP), which is commonly used as a carrier of protein into cells. Arginine-rich CPP has been used as a carrier for the intracellular delivery of various bioactive molecules such as proteins, peptides, and nucleic acids. The number of arginine residues used as CPP can affect the efficiency of cellular uptake and cytosolic release. It is well understood that high cellular uptake efficiency is achieved with 8-12 residues.15 Polyarginine as CPP also plays an essential role in forming hydrogen bonds with cell membrane protein groups.16
Expression of EGFP protein, as inferred from the fluorescence and Western blot data, indicated that the induction of the recombinant cultures had been successful. Analysis performed at 48 and 72 hours post-inoculation to induction medium showed no increase in fluorescent intensity with time (Figure 5B). Thus, in the future, this could be used as a reference supporting that it would only take 48 hours of culturing for expression, hence being more time-efficient.
It is known that the addition of a CPP sequence can have a negative influence on recombinant protein yield.17 It was reported that ten arginines (R10) residues were added to the superfolder GFP (sfGFP) N-terminal in pBAD plasmid containing arabinose-inducible sfGFP gene with a C-terminal His tag. No recombinant R10-sfGFP was expressed by E. coli TOP10. However, the unmodified sfGFP gene expressed over 30 mg/L protein under the same condition.18 It was also reported that although the fluorescent protein obtained after purification showed a single band on SDS-PAGE, mass spectrometry (MS) analysis revealed truncation of the R10, leaving only two arginine residues present at the C-terminus upon expression when the position of R10 and His-tag was swapped relative to the sfGFP gene (i.e. His tag at the N-terminus and R10 at the C-terminus).19
In contrast to these previous findings, our result indicated that the addition of a PolyR group to the C-terminal of EGFP carrying C-terminal 6×His-tag did not seem to interfere with protein expression in S. cerevisiae INVSc1, as shown by the similar fluorescence (Figure 5B) and protein (Figure 6) levels expressed by yeasts carrying pYES2CT-EGFP band pYES2CT-EGFP-PolyR. In combination with the data from the proteins' tertiary structural predictions and alignment, this expression result indicated that the conformational change caused by the addition of PolyR did not affect the protein expression.
We must take into account, however, that the number of arginine residues in PolyR (11×R), the fluorescent gene, and the host used in our study differ from the aforementioned studies by Reddington18 and Patel.19 It would be interesting to observe whether relocating the 6×His-tag or the PolyR to different terminals would improve the protein expression as well as provide ease of purification since it is feared that the PolyR can interfere with the binding of 6×His-tag to Ni-NTA resin to be used for purification or the anti-His-tag antibody used for detection of expression.
The amount of protein expressed can also be influenced by codon preference. The EGFP and EGFP-PolyR encoding genes in this study used codon preference for expression in mammalian cells and had not been adapted for expression in yeast. Analysis of codon adaptation index (CAI) of EGFP-PolyR encoding gene using CAICal20 with S. cerevisiae codon usage table (CUT) from Codon Usage Database showed that the CAI value was 0.575 with GC content 62.3% and Nc 24.4, whereas the EGFP encoding gene had CAI value 0.593 with GC content 60.9% and Nc 27.8.
An alternative that could be attempted to improve the expression is codon optimization. Attempt to optimize the codons encoding the two proteins using ATGme, produced new sequences with better CAI value (0.805), GC content 40.6% and Nc 46.1 for EGFP, and CAI value 0.801, GC content 40.2% and Nc 48.1 for EGFP-PolyR. The optimized sequences are presented in Figure 9.
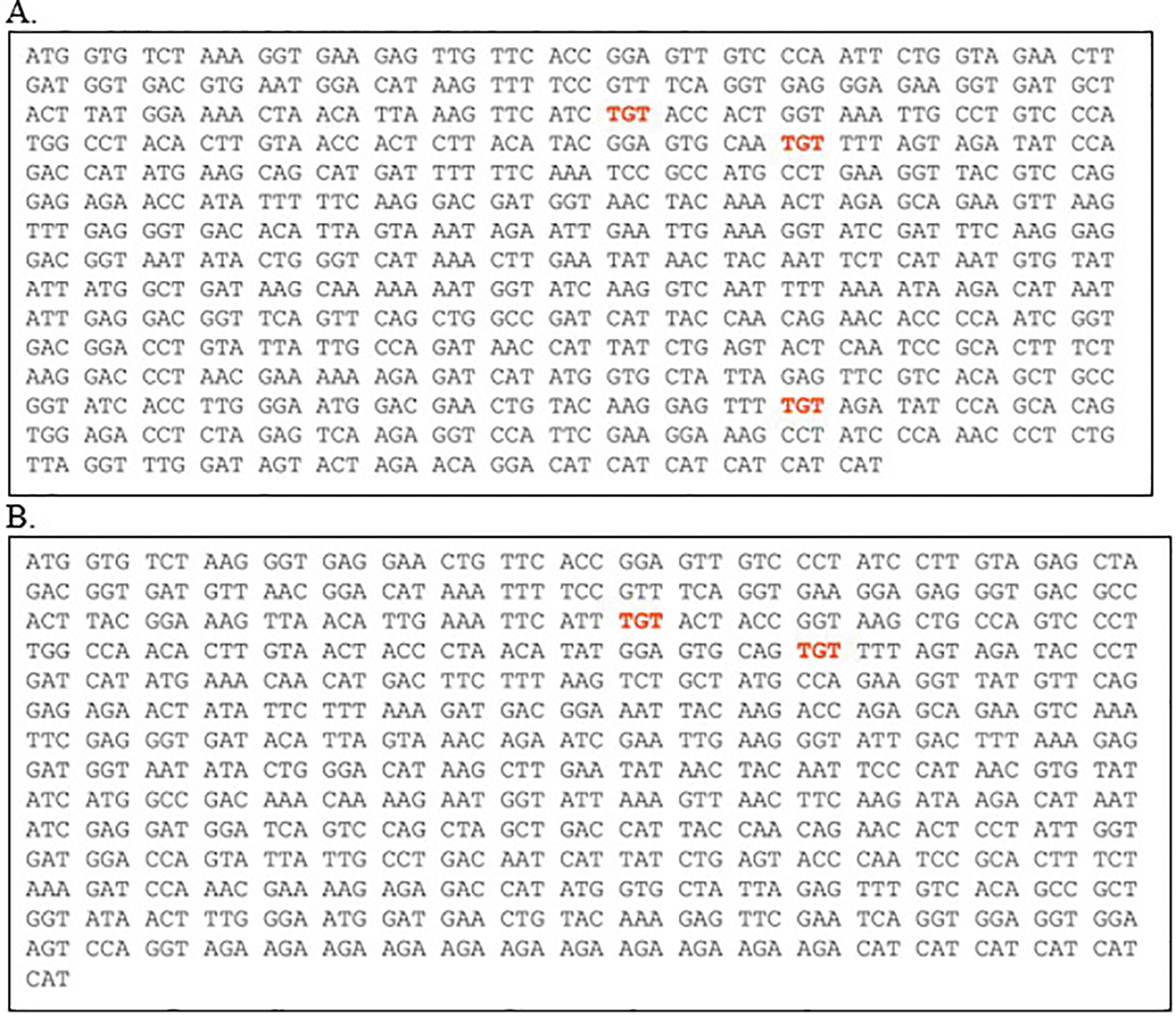
Red indicates codons with frequency <10% in S. cerevisiae.
Nc is the effective number of codons used in a gene, which is calculated from codon usage data, independent of gene length and amino acid (aa) composition.21 Nc values might start from 20, where one codon is exclusively used for each aa, to 61, where all synonymous codons are used with equal frequency.21 CAI measures the deviation of a given protein coding gene sequence with respect to a reference set of genes. In this case, S. cerevisiae CUT was used as a reference. CAI value ranges from 0 to 1, being 1 if a gene always uses the most frequently used synonymous codons in the reference set.20 In terms of gene expression level, generally, a CAI of 1.0 is considered perfect, whereas a CAI of > 0.8 is regarded as good.
During protein synthesis, genetic information encoded in messenger RNAs (mRNAs) is used to direct the synthesis of proteins. This process occurs with great accuracy and speed on the ribosome, using transfer RNAs (tRNAs) as adaptor molecules that carry the amino acids to the mRNA template. For this reason, many argue that designing a gene with CAI 1.0 and Nc 20 is not the best approach to get optimum expression since using only one type or low variations of the codon for each amino acid might overload certain types of tRNAs only, thus blocking or stalling the translation process, hence the protein expression. Therefore, it is sensible to compensate the CAI value to 0.8-1.0 by increasing the Nc value in order to spread the codon usage.
The lower growth curve of the transformant S. cerevisiae compared to the wild-type can be due to the growth inhibition factors of S. cerevisiae activated due to the addition of MM induction medium (2% galactose and 1% raffinose). Adding raffinose to the growth medium of S. cerevisiae can activate the programmed cell death formation of acetic acid. Guaragnella's study (2013) suggests that S. cerevisiae is resistant to programmed cell death via acetic acid because S. cerevisiae can compensate by activating retrograde mitochondria. Although programmed cell death is activated, they are not necessarily inhibited by growth.18 This can be seen in the growth curve of the modified protein transformant. The low growth curve of the transformant S. cerevisiae does not affect the expression of modified proteins (Figures 1 and 2). This is possible because plasmid vectors have been designed to express them in sufficient quantities within the cell.
The S. cerevisiae plasmid vector used in this study is a plasmid vector designed to express its proteins in the cell. This is intended so that the target protein produced is not mixed with other proteins produced as metabolites from S. cerevisiae. Yeast cell walls are relatively rigid structures due to the β(1,3)-glucans.19,20 Depending on the type of acid used for extraction, significantly higher degrees of polymerization have been reported.21 Furthermore, the degree of (1,3)-glucan polymerization may be affected by environmental conditions, as yeast uses two 1,3-glucan synthase complexes to varying degrees depending on the growth phase and carbon source.22 In their mature form, β1,3-glucan chains of stationary phase cells are moderately branched and contain about 3–4% β1,6-linked glucose residues.23 Stationary cell phase 1,3-glucan chains are moderately branched and contain about 3–4 percent 1,6-linked glucose residues in their mature form. The degree of branching of 1,3-glucan may also be affected by growth conditions.
Efficient breakage of cell wall strength-providing components is necessary to remove intracellular compounds effectively.24 Incubation with lyticase in combination with sonication can increase protein yield. This is because lyticase can help break down the cell wall of S. cerevisiae, which is quite strong and rigid. The strength and flexibility structure of the cell wall of S. cerevisiae is composed predominantly of β(1,3)-glucans. The structure of β(1,3)-glucans are influenced by the environment and the nutrients it grows on. The possible addition of an MM induction medium may make the structure of β(1,3)-glucans stronger and difficult to lysis.
The expression results of protein EGFP and its PolyR fused counterpart, EGFP-PolyR indicated that the predicted conformational change caused by PolyR addition in EGFP-PolyR did not affect the expression levels, since both proteins were expressed at an equal level, albeit low. There is the possibility that this low expression level was caused by unoptimized codons used in the gene construct. The construct was not codon optimized for the expression host, S. cerevisiae INVSc1. Codon optimization might be attempted to improve the CAI and Nc values to better match the expression host.
The growth curve of the transformant S. cerevisiae decreased due to the addition of induction mediums, especially raffinose, although it did not affect the amount of expression of the target-modified protein. A sufficient amount of target modified proteins expression for protein isolation in S. cerevisiae by induction of galactose and raffinose can be achieved by using lyticase combined with sonicators in the process of lysis S. cerevisiae. The expression of the modified protein in S. cerevisiae is not affected by the addition of arginine.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)