Keywords
Engineered Heart Tissue (EHT), 3D cardiac model, induced pluripotent stem cell derived cardiomyocytes (iPSC-CM), induced pluripotent stem cell derived cardiac fibroblasts (iPSC-CF), cardiac co-culture, cardiac fibrosis
This article is included in the NC3Rs gateway.
Engineered Heart Tissue (EHT), 3D cardiac model, induced pluripotent stem cell derived cardiomyocytes (iPSC-CM), induced pluripotent stem cell derived cardiac fibroblasts (iPSC-CF), cardiac co-culture, cardiac fibrosis
This is a methods paper describing the generation and incorporation of human induced pluripotent stem cell derived cardiomyocytes and cardiac fibroblasts into engineered heart tissues. The updated version addresses reviewer’s comments. We have calculated and included the contractile force of the engineered heart tissues relative to the cross-sectional area (Figure 6). This exacerbates the differences in contractile function between the engineered heart tissues with and without cardiac fibroblasts. The methods have been appropriately adjusted and the raw data values on the contractile kinetics of the engineered heart tissues have been made available. We have split Figure 7 into two separate figures. The updated Figure 7 demonstrates the alignment of the engineered heart tissues. The channels have been separated in the newly created Figure 8 to clearly demonstrate vimentin and titin immunofluorescence in the engineered heart tissues.
See the authors' detailed response to the review by Emma Louise Robinson
See the authors' detailed response to the review by Mike Dodd
See the authors' detailed response to the review by Claudia Crocini
Scientific benefits
• Generation of a more physiologically relevant human cardiac model
• Utility as a genetically homogenous system for disease modelling of cardiac arrhythmia, heart disease and cardiomyopathy
• Capacity in exploring cellular dynamics integral to cardiac fibrosis
3Rs benefits
• Engineered heart tissue models can replace the use of small animal models of cardiac arrhythmia, cardiomyopathy and heart failure
• Acquiring data from the small animal models used in cardiovascular research often requires the animal to undergo multiple moderate severity procedures which require repeated anaesthesia, such as electrocardiograms and mini pump/telemeter implantation
• As mouse physiology is vastly different to human hearts, insights from mouse models cannot always be extrapolated to human hearts. In contrast, engineered heart tissue models use human cells with human physiology
• Many of the pathogenic variants modelled in mouse lines cause chronic and severe illness
Practical benefits
• The relatively low cost in comparison to mouse models
• Induced pluripotent stem cells are relatively easier to genetically manipulate than in vivo models
• Ability to generate healthy human quiescent cardiac fibroblasts
• Reproducible methods for the derivation of cardiomyocytes and cardiac fibroblasts from hiPSCs
• Accessibility of cardiac cells with a consistent genome
Current applications
• Disease modelling of cardiac arrhythmia, cardiomyopathy and heart failure
• Exploring physiological interplay between cardiac fibroblasts and cardiomyocytes
Potential applications
Limited availability of healthy human heart tissue combined with its inherent nature as a non-proliferative tissue type has meant that heart disease researchers have historically been reliant on in vivo models. Large animal models used in heart disease research, for example dogs, pigs, and sheep, are broadly analogous to humans in cardiac physiology and anatomy but are heavily restricted by cost, throughput, and the sentiment of the general public (Camacho et al., 2016). Small animal models commonly used in cardiovascular research, such as the mouse and zebrafish, offer a cheaper but drastically less physiological and anatomical alternative. Zebrafish offer distinct advantages including optical transparency and rapid development but are limited in their capacity as a cardiac model by their two-chambered heart and temperature-dependent action potential (Vornanen and Hassinen, 2016). Significant differences in the intracellular electrophysiology of mouse cardiomyocytes, caused by contrasting expression and activation of the delayed rectifier and transient outward K+ currents as well as the voltage-gated sodium and calcium channels, are broadly illustrated in their comparatively rapid heart rate of between 500-700 bpm (Xu et al., 1999; Niwa and Nerbonne, 2010; Blechschmidt et al., 2008). Consequently, the modelling of cardiac diseases such as arrhythmia, cardiomyopathy and heart failure, which often manifest through concomitant electrical and structural remodelling, is often hampered in mice due to distinct species-dependent differences.
The co-development of human-induced pluripotent stem cell (hiPSC) technology and CRISPR-Cas 9 genome editing provides a new type of genetically modified disease model based on human cells (Ma et al., 2018; Mosqueira et al., 2018). The utility of induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CM) has caused a shift in the approach many cardiovascular researchers take to cardiac disease modelling, with many groups opting to replace or supplement the use of small animal models with comparatively cheaper stem cell-based models (Cumberland et al., 2022). Consequently, hiPSC-CM have been used to successfully model pathogenic variants that predispose individuals to cardiac diseases, including arrhythmia and cardiomyopathy (Lin et al., 2015; Goktas Sahoglu et al., 2022; Jung et al., 2022; Reyat et al., 2020; Schulz et al., 2023).
The incorporation of hiPSC-CM into cardiovascular disease modelling has however not come without difficulty. Early attempts at differentiating cardiomyocytes produced variable and electrophysiologically immature cells with depolarized diastolic membrane potentials, slow action potential upstroke velocities and large pacemaker currents (Knollmann, 2013). Optimisation and development of differentiation protocols enabled the derivation of chamber-specific cardiomyocytes to explore diverse disease aetiologies but offered marginal advancements in variability and maturity (Jeziorowska et al., 2017; Devalla et al., 2015). Accordingly, innovative maturation strategies were developed to increase the maturity of the cells produced. Current methods include: micropatterning, metabolic maturation, electrical stimulation, soft-substrate culture, co-culture, and engineered 3D models (Jimenez-Vazquez et al., 2022; Feyen et al., 2020; Yoshida et al., 2018; Tzatzalos et al., 2016; Machiraju and Greenway, 2019).
With greater electrophysiological, metabolic, structural, and functional maturity, 3D cardiac models offer greater potential in understanding the nuances of complex cardiac diseases such as heart failure and cardiomyopathy (Correia et al., 2018; Shadrin et al., 2017; Ulmer et al., 2018; Vučković et al., 2022). Furthermore, the concurrent ease in which contractile force and transmembrane potential can be monitored in 3D cardiac models presents great potential for their future incorporation into in vitro cardiotoxicity studies (Gintant et al., 2019). A number of 3D cardiac models have been generated to examine cardiac physiology in disease including, but not limited to: two-post engineered heart tissues (EHTs), ring-shaped engineered heart muscle (EHM), cardiac patches, cardiac micro-, and biowires, and Novoheart (Tiburcy et al., 2017; Ronaldson-Bouchard et al., 2018; Zhang et al., 2013; Thavandiran et al., 2013; Zhao et al., 2019). Although differing substantially in size and shape, all the 3D cardiac models listed above attempt to better recapitulate the cell-to-cell and cell-to-matrix interactions present in the myocardium and encourage the longitudinal alignment of cardiomyocytes upon a scaffold (Smith et al., 2017).
Despite occupying the majority of the volume of the mammalian heart, cardiac myocytes are estimated to account for less than 50 % of the total cell number (Zhou and Pu, 2016). Major non-myocyte cell types in the heart include cardiac fibroblasts and endothelial cells as well as ancillary populations of immune cells and autonomic neurones. Therefore, the incorporation of non-myocyte cell populations into cardiac modelling systems should facilitate the structural, metabolic, and electrophysiological maturity of hiPSC-CM and aid in generating a more physiologically relevant microenvironment (Yoshida et al., 2018; Kim et al., 2010). Up to now, cell types commonly incorporated into 3D cardiac models include endothelial cells, mesenchymal stem cells, and cardiac fibroblasts (Tadano et al., 2021; Tulloch et al., 2011; Zhang et al., 2017).
Cardiac fibroblasts are a common cell type in human ventricles (circa 20 % by number, [Pinto et al., 2016]) and are integral to the architecture, alignment, and electromechanical properties of the myocardium in health and disease. Quiescent or inactive induced pluripotent stem cell-derived cardiac fibroblasts (hiPSC-CF) can be derived from hiPSCs and used effectively as a stand-alone model of cardiac fibrosis (Zhang et al., 2019). The trans-differentiation of the quiescent cardiac fibroblast to myofibroblast occurs prior to and during the development of cardiac fibrosis and is often difficult to prevent in vitro as the cells cultured on plastic dishes are subject to a Young’s modulus up to a million times stiffer than the native myocardium (Landry et al., 2019; Acevedo-Acevedo and Crone, 2015). This pathological transition is substantially stimulated by the profibrotic signalling cytokine TGF-β1. Suppression of TGF-β1 can be achieved in vitro using small molecule inhibitors such as SB 431542 and can be used on in vitro models to control the activation status of quiescent cardiac fibroblasts (Law and Carver, 2013).
An amalgamated 3D cardiac model consisting of hiPSC-CF and hiPSC-CM presents the opportunity for the generation of genetically homogenous (isogenic) systems capable of investigating the pathophysiological interplay present in cardiac diseases such as cardiomyopathy and arrhythmia. Here we describe a modified protocol for the generation and incorporation of quiescent hiPSC-CF into an engineered heart tissue model.
In this study we describe efficient protocols for the differentiation of hiPSC-CM and quiescent cardiac fibroblasts. Methods are outlined detailing the incorporation of these cells into EHTs with improved contractile function and tissue compaction and potential use exploring the pathophysiological interplay between hiPSC-CM and hiPSC-CF in cardiac fibrosis. In the UK, 51,427 procedures were carried out on mice in 2021 for research on the cardiovascular, blood and lymphatic system (Home Office Report on Annual Statistics of Scientific Procedures on Living Animals Great Britain 2021). Assuming that 5 % of these mice were used to study cardiac arrhythmia, heart failure or cardiomyopathy, use of the model proposed could lead to the direct replacement of 2500 mice per year in the UK alone.
Induced pluripotent stem cells Kolfc2 (WTSIi018-B) were maintained on six-well plates coated with Geltrex (1:100; for details of reagents and suppliers see Table 1) according to the manufacturer’s protocol. Cells were cultured in mTeSR Plus media at 37 °C and 5 % CO2 and passaged at 70-80 % confluency. Cells were passaged by removing the media in the well, washing once with PBS and adding TrypLE Express Enzyme (1X), phenol red for 3 minutes at 37°C. The TrypLE Express was removed and the cells were gently washed off the surface of the well with 1 mL of warm mTeSR Plus medium containing 10 μM Rock Inhibitor (Y-27632). Cells were passaged according to a seeding density of 20,000 cells per cm2 and cultured in 10 μM Rock Inhibitor (Y-27632) mTeSR Plus media for the first 24 hours before switching to mTeSR Plus. The Rho-kinase inhibitor (Rock Inhibitor), Y-27632 was added to prevent dissociation-induced apoptosis. It should be removed 24 hours after the passaging of the cells to maintain iPSC pluripotency.
| Reagent and preparation | Company and catalogue number |
|---|---|
| Kolfc2 (WTSIi018-B) | https://ebisc.org/WTSIi018-A |
| mTeSR Plus | StemCell Technologies, 100-0276 |
| DPBS, no calcium, no magnesium | ThermoFisher Scientific, 14190144 |
| Corning Costar 6-well Clear TC-treated | Fisher Scientific, 10578911 |
| Corning 25 cm2 cell culture flask TC-treated rectangular canted neck | Sigma-Aldrich, CLS430639-200EA |
| Geltrex LDEV-Free Reduced Growth Factor Basement Membrane Matrix | ThermoFisher Scientific, A1413201 |
| TrypLE Express Enzyme (1X), phenol red | ThermoFisher Scientific, 12605010 |
| TrypLE Select Enzyme (1X), no phenol red | ThermoFisher Scientific, 12563011 |
| Rock Inhibitor Y-27632 (Dihydrochloride) (Reconstituted in PBS to 10 mM and stored at -20 °C in 50 μL aliquots) | StemCell Technologies, 72304 |
| StemPro-34 SFM (1X) | ThermoFisher Scientific, 10639011 |
| L-Glutamine (200 mM) | ThermoFisher Scientific, 25030081 |
| Recombinant Human BMP-4 Protein (Reconstituted in 4 mM HCl (0.1 % BSA) to 50 μg/mL and stored at -80 °C in 5 μL aliquots) | R&D Systems, 314-BP-010/CF |
| Human Activin A Recombinant Protein (Reconstituted in PBS (0.1 % BSA) to 10 μg/mL and stored at -20 °C in 20 μl aliquots) | ThermoFisher Scientific, PHC9564 |
| RPMI 1640 Medium | ThermoFisher Scientific, 11875093 |
| B-27 Supplement, minus insulin | ThermoFisher Scientific, A1895601 |
| XAV 939 (Reconstituted in DMSO to 10 mM and stored at -20 °C in 25 μL aliquots) | TOCRIS, 3748 |
| KY 02111 (Reconstituted in DMSO to 10 mM and stored at -20 °C in 25 μL aliquots) | TOCRIS, 4731 |
| B-27 Supplement (50X), serum free | ThermoFisher Scientific, 17504044 |
| Retinoic Acid (Reconstituted in DMSO to 50 mM and stored at -80 °C in 5 μL aliquots) | Sigma-Aldrich, R2625-50MG |
| RPMI 1640 Medium, no glucose | ThermoFisher Scientific, 11879020 |
| CHIR 99021 (Reconstituted in DMSO to 10 mM and stored at -20 °C in 10 μl aliquots) | TOCRIS, 4423 |
| IWR-1 (Reconstituted in DMSO to 10 mM and stored at -20 °C in 10 μL aliquots) | Sigma-Aldrich, I0161 |
| Accutase solution | Sigma-Aldrich, A6964 |
| Advanced DMEM/F-12 | ThermoFisher Scientific, 12634010 |
| Fibroblast Growth Medium 3 | (PromoCell, C-23130) |
| Recombinant Human FGF basic/FGF2/bFGF (146 aa) Protein (Reconstituted in PBS (0.1 % BSA) to 10 μg/mL and stored at -80 °C in 50 μL aliquots) | bio-techne, 233-FB-010 |
| SB 431542 (Reconstituted in DMSO to 10 mM and stored at -80 °C in 50 μL aliquots) | TOCRIS, 1614 |
The method for differentiation of hiPSC-CM was broadly adapted from the protocol outlined in Smith et al. (2018) (Figure 1). hiPSCs were seeded onto six-well plates coated with Geltrex and cultured in 2 mL per well of mTeSR Plus. The medium was changed on the cells every 48 hours until the cells reached 60 % confluency. The medium was then changed for 24 hours with StemPro-34 SFM (1X) supplemented with 2 mM L-Glutamine, 1 ng/mL Recombinant Human BMP-4 Protein and 1:100 Geltrex. The cells were subsequently changed with StemPro medium supplemented with 10 ng/mL BMP-4, 8 ng/mL Activin A and 2 mM L-Glutamine and incubated for 48 hours (day 0). The medium was changed with RPMI 1640 Medium with B-27 Supplement, minus insulin, 10 μM XAV 939 and 10 μM KY 02111 for 48 hours (day 2). The cells were subsequently changed with RPMI 1640 Medium with B-27 Supplement (50X), 10 μM XAV 939 and 10 μM KY02111 for 48 hours (day 4). The medium on the cells was changed with RPMI 1640 Medium with B-27 Supplement (50X) every other day. Atrial differentiation was achieved through the addition of 1 μM retinoic acid to the media on days 4 and 6. At 12 days after the initiation of differentiation, glucose starvation was performed to purify the population of cells for cardiomyocytes. This was achieved by changing the medium of the cells with RPMI 1640 Medium supplemented 1:50 with no glucose with B-27 Supplement (50X) for 48 hours. Cells were dissociated at day 15 for incorporation into EHT models. This media was removed from the well(s), the well was washed once with 1 mL of PBS and the cells were incubated in 2 mL of TrypLE Select 10× for 30 minutes at 37 °C. The cells were then washed from the surface of the plate with pre-warmed DMEM-F12.
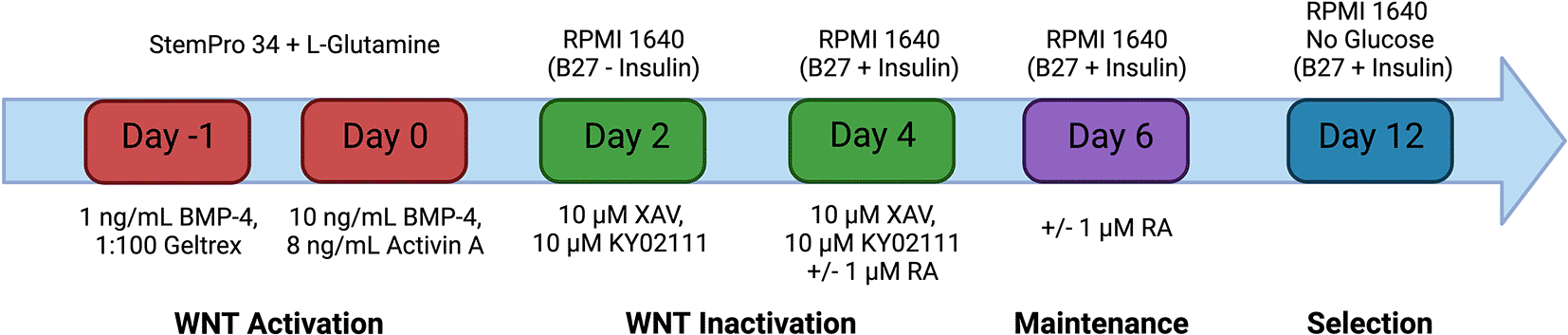
Schematic representation of the hiPSC-CM differentiation protocol. Mesodermal specification is achieved through the activation of the Wnt signalling pathway using the recombinant proteins: Bone morphogenic protein- 4 (BMP-4) and Activin A. Subsequent inactivation of the Wnt signalling pathway by small molecules XAV 939 and KY02111 enables the differentiation of cardiac progenitor cells into induced pluripotent stem cell derived cardiomyocytes. Differentiation can be driven towards atrial cardiomyocyte specification through the addition of retinoic acid on days 4 and 6 only. The medium on the cells was changed every 48 hours after day 6 with RPMI 1640 with B27 + Insulin. Selection is achieved from days 12-14 by culturing the cells in RPMI 1640 No Glucose with B27 + insulin for 48 hours. Created with Biorender.com.
The method for the differentiation of quiescent induced pluripotent stem cell-derived cardiac fibroblasts was broadly adapted from the protocol outlined in Zhang et al. (2019) (Figure 2). Induced pluripotent stem cells (Kolfc2/WTSIi018-B) were seeded at 30,000 cells/cm2 in a T25 flask pre-coated with 1:100 Geltrex for 2 hours at 37 °C, in 10 μM Rock Inhibitor (Y-27632) mTeSR Plus medium. The cells were cultured for 48 hours prior to the medium being changed with RPMI 1640 supplemented with B27 minus insulin and 6 μM CHIR-99021 for 48 hours. The medium was then changed with RPMI 1640 supplemented with B27 minus insulin for 24 hours. The cells were subsequently treated with a RPMI 1640 with B27 minus insulin and 5 μM IWR1 for 48 hours. The cells were dissociated by washing with 3 mL PBS and then treating with 5 mL accutase enzyme for 3 minutes at 37 °C. The cells were removed from the flask, diluted with 5 mL pre-warmed RPMI 1640 and spun down at 200 g for 3 minutes. The cells were resuspended in Advanced DMEM/F-12 supplemented with 5 μM CHIR-99021 and 2 μM Retinoic Acid and seeded at 20,000 cells/cm2 in a Geltrex coated T25 flask. The cells were incubated in this medium for 3 days and then subsequently changed to Advanced DMEM/F-12 for a further 4 days. The cells were passaged and plated into a Geltrex coated T25 flask at a density of 20,000 cells/cm2 in Fibroblast Growth Medium 3 supplemented with 10 ng/mL FGF2 and 10 μM of the TGF-β1 inhibitor SB431542. The cells were cultured in this media for 6 days, with media changes taking place every 48 hours. Following the 6 days, the cells can be assessed for purity using immunocytochemistry. It is recommended that the cells are expanded, passaged at 70-80 % confluency for 2 passages and cryopreserved for use in future experiments in serum free freezing medium. Cells were passaged by washing once with PBS, and then incubating in TrypLE™ Express for 3 minutes at 37 °C. Cells can be dislodged from the surface of the flask by gently tapping the sides of the container. Following the detachment of the cells, a 2 × volume of DMEM/F12 was added to dilute the TrypLE. The TrypLE/cell mix was centrifuged at 300 g for 3 minutes. The cells were resuspended in Fibroblast Growth Medium 3 supplemented with 10 ng/mL FGF2 and 10 μM SB431542 and seeded at a density of 10,000 cells/cm2.
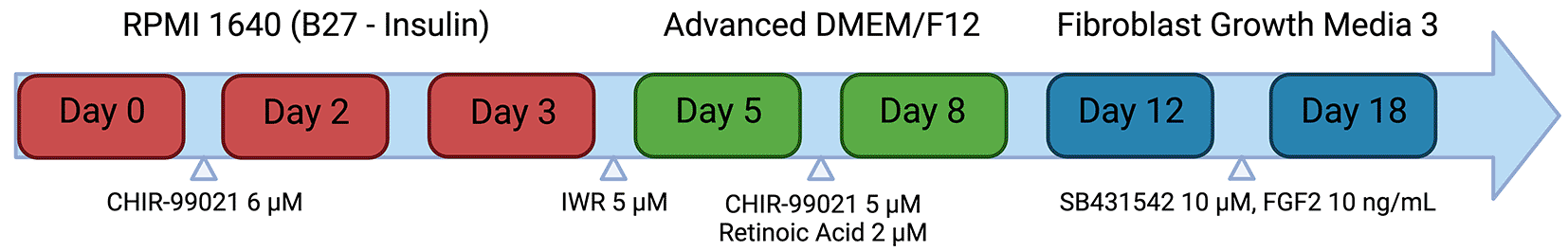
Schematic representation of the hiPSC-CF differentiation protocol. Mesodermal specification is achieved through the activation of the Wnt signalling pathway using CHIR-99021. Subsequent inactivation of the Wnt signalling pathway by the small molecule IWR1 allows the generation of cardiac progenitor cells. Culture in CHIR-99021 and retinoic acid drives the cells towards the derivation of cardiac fibroblasts. Cardiac fibroblasts are maintained in medium supplemented with FGF2 and the TGF-β1 inhibitor SB 431542 to prevent activation into myofibroblasts. Created with Biorender.com.
Protocol for EHT construction
Here we describe a step-by-step protocol for the construction of four EHTs (one strip) containing iPSC-CF. The reagents required are listed in Table 2.
1. Remove media and wash 1-2 confluent wells of a six-well plate containing iPSC-CM with room temperature PBS once
NOTE: 1 × confluent well of hiPSC-CM in a 6 well plate should yield approximately 4 × 106 cells
2. Add 2 mL of TrypLE Select Enzyme (10X) per well of a 6 well plate and incubate for 30 minutes at 37 °C
3. Transfer the detached cells to a 50 mL centrifuge tube and wash off any attached cells with 1 mL per well of pre-warmed DMEM-F12
4. Add DMEM-F12 to the cell mix to give a total volume of 10 mL
NOTE: Centrifugation of iPSC-CM takes place later, when both cell types have been combined
5. Perform a cell count
1. Remove the media and wash a T25 flask (80 % confluency) containing iPSC-CF with PBS
NOTE: A T25 flask at 80 % confluency should yield approximately 600,000 iPSC-CF
2. Add 3 mL TrypLE Express and incubate for 3 minutes at 37 °C
3. Gently tap the flask on all sides to dissociate cells from the surface of the flask
4. Transfer the 3 mL of TrypLE/cell mix to a 50 mL centrifuge tube
5. Rinse the flask with 7 mL of pre-warmed DMEM-F12 before combining it with the TrypLE containing cells
6. Perform a cell count
1. Combine required volumes of hiPSC-CM (4 × 106 cells) and hiPSC-CF (6 × 105 cells) cell mixes in a centrifuge tube and make up to 30 mL with pre-warmed DMEM-F12
2. Spin down the tube containing both iPSC-CM and iPSC-CF at 100 g for 10 minutes
3. Place thrombin, Fib-Ap, Rock Inhibitor and Geltrex on ice
4. Following centrifugation, gently place the cells on ice
NOTE: Be careful not to disturb the pellet
1. Microwave 2 % agarose until completely molten
2. Pipette 1.6 mL of molten agarose per well in a column of a Nunc 24 well plate
3. Insert a Teflon spacer into a column of the Nunc 24 well plate containing molten agarose to create agarose moulds for a silicon rack of four EHTs
NOTE: Wells of a 24 well plate can differ in dimensions. The spacers and silicon racks are optimised for use in Nunc Cell-Culture Treated 24 well plates
4. Remove the Teflon spacers from the now solidified agarose moulds after 15 minutes
NOTE: Agarose moulds can deteriorate and crack over time. Cast EHTs in the agarose moulds 10 minutes after removal of the Teflon spacers
5. Place the silicon racks into the moulds, ensuring central alignment
6. NOTE: Misaligned silicon racks increase the risk of snagging during removal from the moulds
1. Aliquot 3 μL of the thrombin into 4 × 0.5 mL microcentrifuge tubes and chill on ice (4 tubes for 4 EHTs)
NOTE: Briefly spin down the tubes containing the thrombin
2. Add 368.55 μL of NKM medium to a prechilled 1.5 mL microcentrifuge tube
3. Prewarm the Fib-Ap mix in your hands (to enable accurate pipetting) and then add 11.25 μL to the NKM medium
NOTE: Complete dissolution of the Fib-Ap mix is paramount to successful generation of EHTs.
4. Add 24.75 μL of 2X DMEM
5. Add 0.45 μL of Rock Inhibitor
6. Add 45 μL of geltrex
7. Resuspend the cell pellet containing both hiPSC-CM and hiPSC-CF in the EHT mix
NOTE: Everything should be kept on ice at this stage.
1. Add 100 μL of the EHT cell mix to a 3 μL aliquot of thrombin and then quickly pipette the mix in between the two silicon posts resting inside the agarose mould
NOTE: Minimise risk of air bubble formation by only pipetting down to the first stop of the pipette
2. Repeat step 1 for each EHT
NOTE: Be careful not to knock or disturb the 24 well plate holding the solidifying EHTs
3. Gently triturate the EHT cell mix intermittently (every 4 EHTs) to ensure homogeneity
4. Gently place the lid on the 24 well plate and place in a 37 °C incubator for 2 hours
1. Warm NKM medium in a 37 °C water bath
2. Add 1.5 mL of EHT medium to each well of a column of wells in a Nunc 24 well plate and place in the 37 °C incubator
3. After 2 hours, gently pipette 350 μL of prewarmed NKM medium on top of the agarose mould in each well
NOTE: Pipetting on top of the agarose mould prevents disruption to the EHT
4. Place in the incubator for a further 10 minutes
5. Move the plate from side to side to encourage dislodging of the EHTs
6. Carefully but firmly remove the EHT strips from the agarose moulds and place into the wells containing the prewarmed EHT medium
NOTE: Removal of the strips should be performed in a single action as prolonged and gentle removal of the EHTs from the agarose moulds can increase the risk of snagging and malformation
8. Maintain the EHTs by transferring the silicon racks into wells containing fresh EHT medium every 48 hours.
hiPSC-CF were cultured in T25 flasks for 72 hours in Fibroblast Growth Media 3 supplemented with 10 ng/mL FGF2 + 10 μM SB 431542 or 10 ng/mL FGF2 + 10 ng/mL TGF-β1 (R&D Systems, 240-B-002/CF). RNA extraction was performed using the Direct-zol RNA Miniprep Kit (Zymo Research, R2050) according to the manufacturer’s protocol. cDNA was generated using the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific, 4368814) according to the manufacturer’s protocol. RT-qPCR was performed using TaqMan Fast Advanced Master Mix (ThermoFisher Scientific, 4444557) following the manufacturer’s protocol and on a QuantStudio 5 Real-Time PCR System, 384-well (ThermoFisher Scientific, A28140). RT-qPCR was performed using TaqMan Gene Expression Assay (FAM) (ThermoFisher Scientific, 4331182) for ACTA2 (Hs00426835_g1) and COL1A1 (Hs00164004_m1). All reactions were multiplexed and normalised to the housekeeping gene GAPDH using Human GAPD (GAPDH) Endogenous Control (VIC/MGB probe, primer limited) (ThermoFisher Scientific, 4326317E). 10 ng of cDNA were used per reaction. Transcript levels were measured from three samples of iPSC-CF, with each sample being obtained from cells at different passages. Cycle threshold values were obtained from three technical replicates of each sample. Relative gene expression was calculated using the comparative cycle threshold method (Schmittgen and Livak, 2008).
Induced pluripotent stem cell derived cardiac fibroblasts
hiPSC-CF were split into wells of a Geltrex-coated 24 well plate (10,000/well) and cultured for 72 hours in Fibroblast Growth Media 3 supplemented with 10 ng/mL FGF2 + 10 μM SB 431542 or 10 ng/mL FGF2 + 10 ng/mL TGF-β1. Cells were fixed for 14 minutes at room temperature using 4 % paraformaldehyde Solution (ThermoFisher Scientific, J19943.K2). Fixed cells were blocked for 1 hour in blocking buffer consisting of PBS, Fetal bovine serum (5 %), Bovine Serum Albumin (1 %) (Merck Millipore, A9418-10G) and Triton X-100 (0.5 %) (Merck Millipore, X100-100ML). Primary antibodies were added at dilutions listed in Table 4 in blocking buffer for 2 hours at room temperature. This was followed by 3 washes in blocking buffer and the addition of secondary antibodies (Table 4) for 1 hour in the dark at room temperature. DAPI (ThermoFisher Scientific, D1306) was added with the secondaries at 0.1 μg/mL to visualise nuclei. Samples were washed 3 times in blocking buffer and then once with PBS. Samples were imaged using a EVOS M5000 Imaging System (ThermoFisher Scientific, AMF5000).
| Target | Company, Catalog | Host | Concentration |
|---|---|---|---|
| Vimentin (Primary) | Abcam, ab45939 | Rabbit | 1:500 |
| α-Smooth Muscle Actin (Primary) | Merck Millipore, A5228 | Mouse | 1:500 |
| Collagen 1 (Primary) | Abcam, ab34710 | Rabbit | 1:500 |
| Titin (T12) (Primary) | Fürst et al. (1988) | Mouse | 2 μg/mL |
| Alexa Fluor 488 anti-Mouse IgG (Secondary) | ThermoFisher Scientific, A28175 | Goat | 1:500 |
| Alexa Fluor 546 anti-Rabbit IgG | ThermoFisher Scientific, A-11035 | Goat | 1:500 |
| DAPI (4′,6-Diamidino-2-Phenylindole, Dihydrochloride) (Nuclear Stain) | ThermoFisher Scientific, D1306 | N/A | 0.1 μg/mL |
Engineered heart tissues
EHTs constructed with and without the addition of hiPSC-CF were removed from posts after 15 days in culture (30 days post initiation of differentiation), washed three times in PBS and fixed overnight at 4 °C in 4 % paraformaldehyde solution. A scalpel was used to longitudinally section EHTs. Sectioned pieces of EHTs were blocked overnight in blocking buffer. Primary antibodies were added in blocking buffer, at the dilutions listed in Table 4, overnight at 4°C. Samples were washed three times in blocking buffer before the addition of secondary antibodies for 4 hours in the dark at room temperature. DAPI was added with the secondaries at 0.1 μg/mL to visualise nuclei. Tissue sections were placed onto coverslips (VWR, 631-0170) and mounted onto cover slides using Hydromount Histology Mounting Media (Scientific Laboratory Supplies, NAT1324). Images were acquired using a Zeiss LSM780 Confocal laser scanning microscope equipped with a C-Apochromat 63x/1.20 W objective.
EHTs constructed with and without hiPSC-CF were cultured for 15 days in EHT media (30 days after initiation of differentiation) prior to recording and analysis. 10 second videos of each EHT was recorded at 1080p/30 fps using an inverted camera with a macroscopic lens (3 EHTs with iPSC-CF, 3 EHTs without iPSC-CF). Videos were converted into tif files and analysed using the ImageJ Plugin TrackMate for displacement over time. All EHTs with contractile function were included in the analysis. Contractile function was defined as at least two macroscopically visible contractions within 15 seconds. All EHTs constructed with iPSC-CF demonstrated contractile function. Only three of the nine EHTs constructed (one of three batches, three EHTs per batch) without the addition of iPSC-CF showed contractile function. The analysis of contractile function was performed blinded to the presence or absence of hiPSC-CF. Outputs were semi-automated and included contraction duration, time to peak, relaxation time, total contraction amplitude and beat rate. Contraction amplitude/area (tensile strength) was calculated by dividing the total contraction amplitudes by the cross-sectional area of the EHTs. For converting displacement into force, elastic bending of the pillars was assumed with a measured Young’s modulus of 3.0 MPa, yielding a conversion factor of 0.26 mN/mm.
Induced pluripotent stem cells were differentiated into cardiac fibroblasts using the protocol outlined in materials and methods. Following differentiation, the activation status and plasticity of the cells was assessed using immunofluorescence. The cells were cultured in the presence and absence of 10 ng/mL TGF-β1 for 72 hours. The cells were then fixed and stained for vimentin, αSMA and collagen 1 with the results shown in Figure 3A. Vimentin is a cardiac fibroblast marker irrespective of activation status whilst αSMA and collagen are predominantly expressed in activated or myofibroblasts. The hiPSC-CF cultured in the absence of TGF-β1 were positive for vimentin but negative for αSMA, indicating a quiescent cardiac fibroblast phenotype. The cells cultured without the TGF-β1 inhibitor, SB 431542, and in the presence of 10 ng/mL TGF-β1 were positive for vimentin and αSMA and showed greater expression of collagen. The gene expression of ACTA2 (the transcript which encodes αSMA) and COL1A1 (a transcript which encodes collagen) was assessed using RT-qPCR as markers for cardiac fibroblast activation. Expression analysis was performed on three biological samples, with three technical repeats performed for each sample. No data were excluded from the analysis. The results are shown in Figure 3B. The hiPSC-CF showed a three-fold increase in the mean expression of COL1A1 following TGF-β1 treatment (-TGF-β1: M = 1, SD = 0.21, +TGF-β1 M = 2.9, SD = 0.47). The mean expression of ACTA2 in the hiPSC-CF increased approximately five-fold following TGF-β1 treatment (TGF-β1: M = 1, SD = 0.13, +TGF-β1 M = 4.9, SD = 1.59).
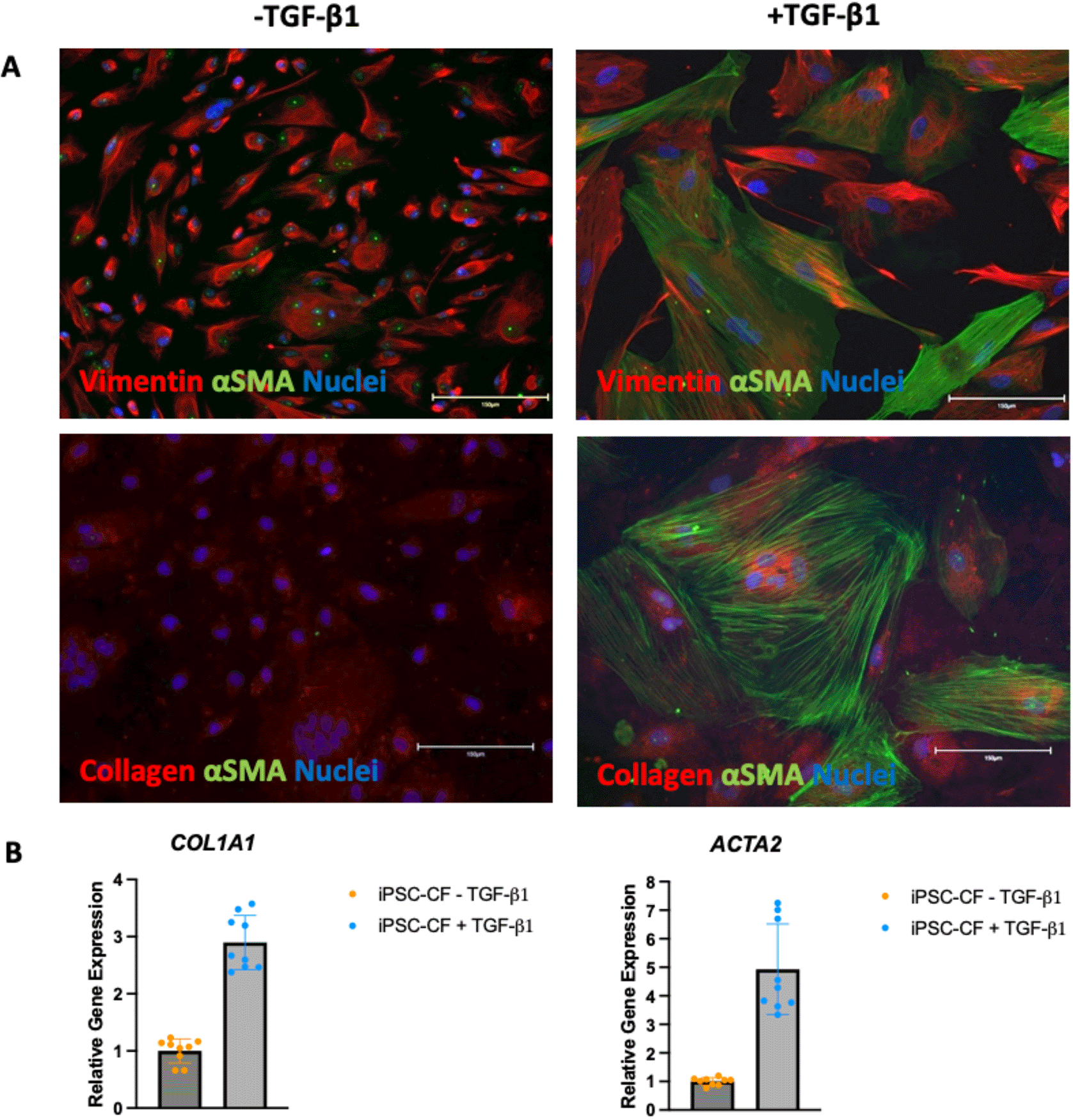
hiPSC-CF were cultured in the standard media (FGM3 + 10 ng/ml FGF2 + 10 μM SB 431542, ‘-TGF-β1’) and with 10 ng/mL TGF-β1 instead of the SB 431542 (‘+TGF-β1’) for 72 hours to assess activation status. A: The cells were fixed and stained for vimentin (top red), Collagen (bottom red) αSMA (green) and DAPI (blue, indicating nuclei). Scale bar = 150 microns. B: RNA was extracted, and RT-qPCR was performed on the cDNA for ACTA2 and COL1A1.
N = 3 (three biological samples with 3 technical replicates per sample are plotted).
A workflow delineating the generation of EHTs from hiPSCs is displayed in Figure 4. EHTs containing hiPSC-CM and hiPSC-CF started twitching approximately 48 hours after construction, with maximal beating capacity being reached after approximately 14 days in culture. Those generated in the absence of hiPSC-CF started twitching approximately 96 hours after construction and showed diminished contractile function throughout the duration of culture. EHTs constructed with and without hiPSC-CF demonstrated marked differences in shape and structure after 72 hours in culture as shown in Figure 5. EHTs constructed with hiPSC-CF showed tightening around the silicon posts whilst hiPSC-CM-only EHTs remained more block-like throughout time in culture.

An outline of the workflow to generate of EHTs with and without the addition of hiPSC-CF. The EHT shown on the top right was generated without the addition of hiPSC-CF. The EHT shown on the bottom right was constructed with the addition of hiPSC-CF. Created with Biorender.com.
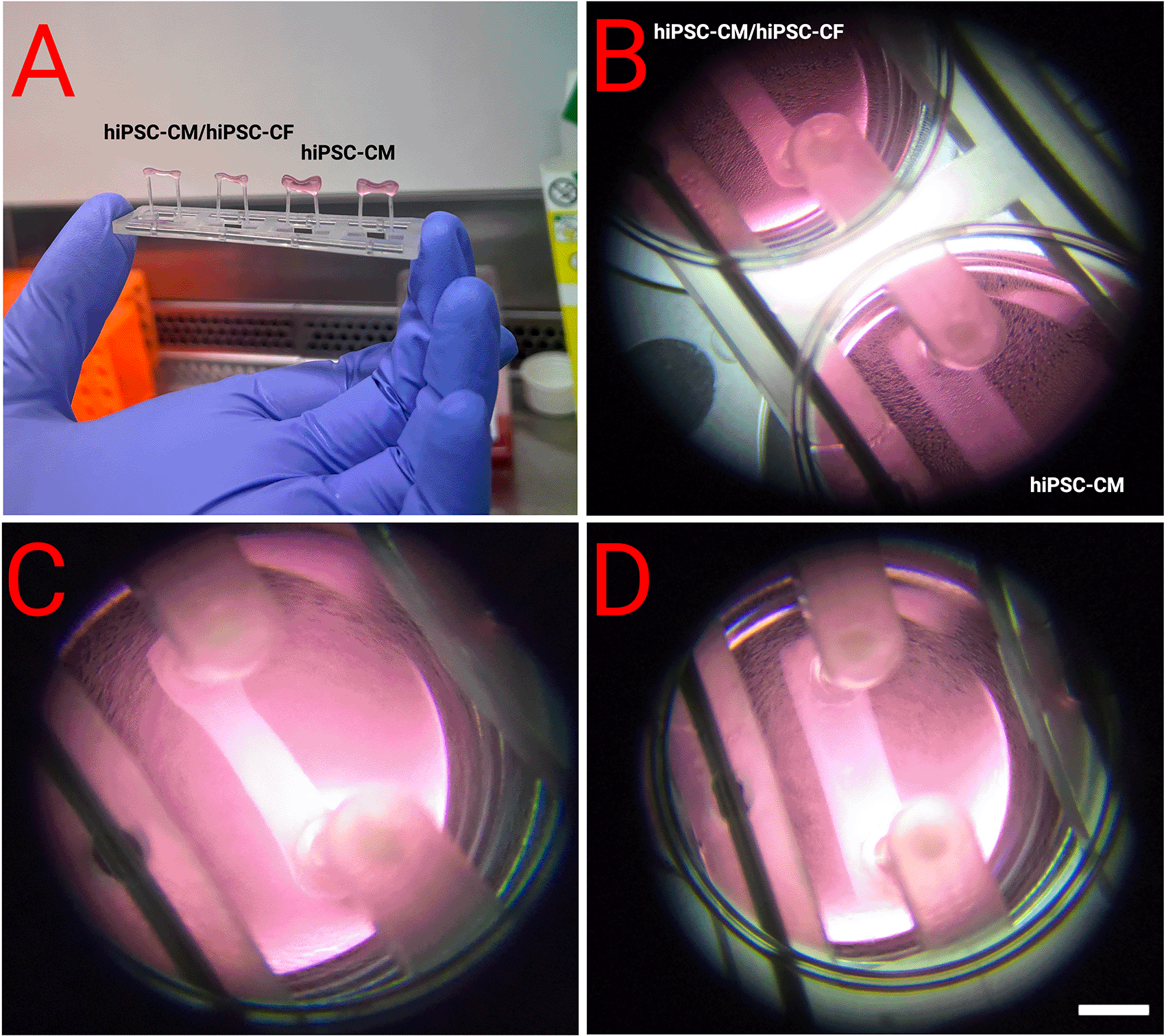
EHTs were constructed with and without the addition of hiPSC-CF according to the protocol outlined above. A and B show side by side comparisons. C - Close-up of an EHT with hiPSC-CF and hiPSC-CM demonstrated compaction after 72 hours, which was not observed in EHTs generated from hiPSC-CMs alone (D). Scale bar = 4 mm.
At day 30 after the initiation of differentiation (15 days in EHT form), 1080p/30 fps videos of EHTs with and without hiPSC-CF were analysed using the Open-source ImageJ plugin TrackMate (Figure 6). All EHTs which demonstrated contractility were included in the analysis (Section 3.8). EHTs with and without hiPSC-CF demonstrated similar contraction durations and relaxation times of approximately 400 and 240 ms respectively. The mean time to peak time of the EHTs containing hiPSC-CF was similar to that of their hiPSC-CM-only counterparts at approximately 170 ms. The average contraction amplitude of the EHTs containing hiPSC-CF was approximately 30 % greater than that of the EHTs consisting of hiPSC-CM only. The EHT containing the hiPSC-CFs demonstrated an approximate five-fold increase in force, normalised to the cross-sectional area. EHTs constructed with hiPSC-CFs showed a marked decrease in spontaneous beat rate.
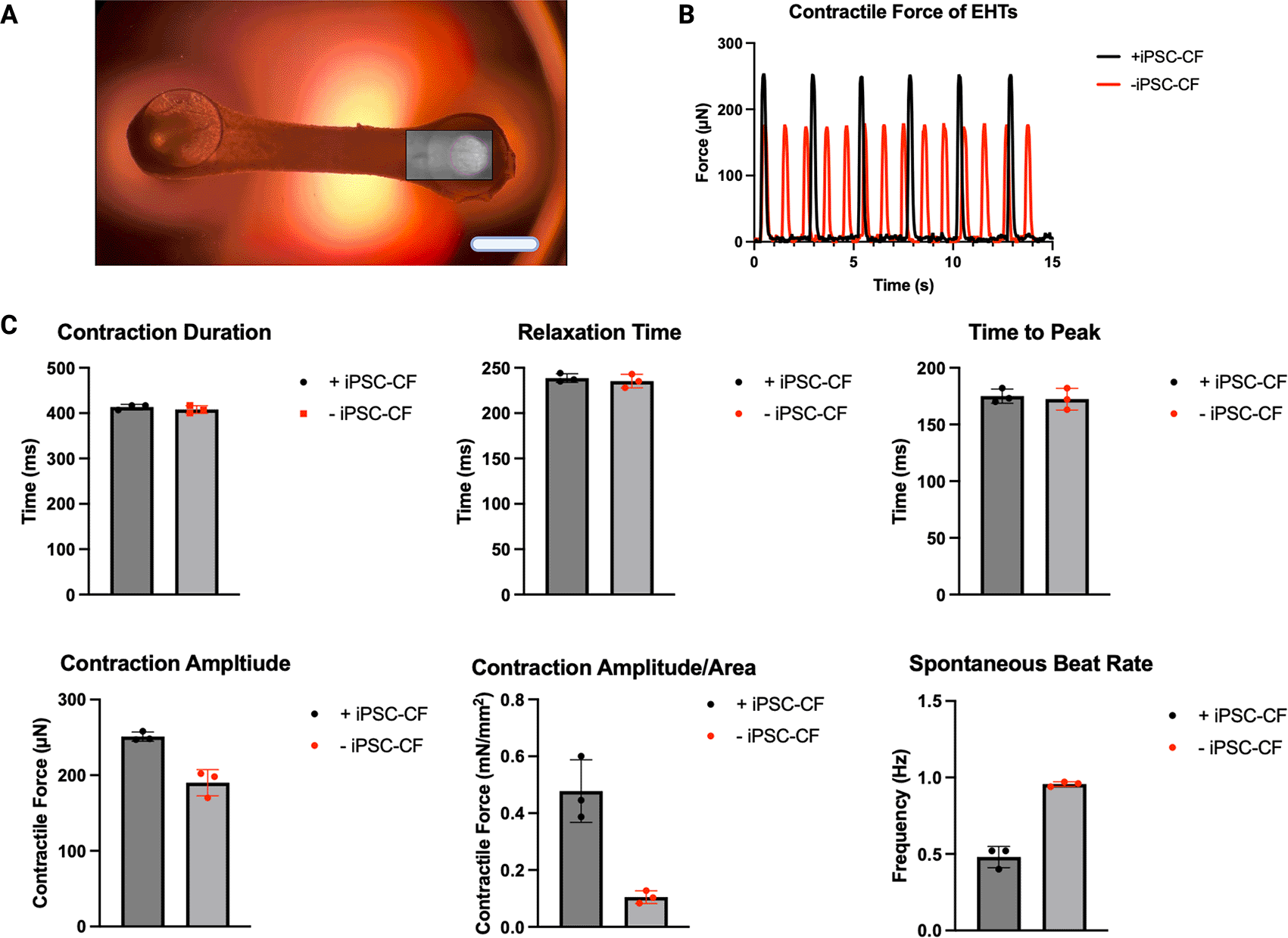
EHTs constructed with and without the addition of hiPSC-CF were analysed to assess contractile activity. Frames of the videos of the EHTs were analysed using the ImageJ Plugin TrackMate for displacement over time, the box represents the analysis window where movement of the pillar was tracked (A). Contractile profiles were generated (a representative profile of an EHT with and without iPSC-CF is shown) (B) and used to ascertain Contractile Duration, Relaxation Time, Time to Peak, Contraction Amplitude, Contraction Amplitude/Area and Spontaneous Beat Rate (C). N = 3 EHTs. Scale bar = 4 mm.
EHTs consisting of hiPSC-CM and hiPSC-CM/hiPSC-CF were cultured for 15 days after construction (30 days after initiation of hiPSC-CM differentiation). The tissue was fixed and sectioned using a scalpel and then stained for DAPI, titin and vimentin to assess composition and alignment (Figures 7, 8). Vimentin is a key marker of cardiac fibroblasts whilst titin is an integral component of the sarcomeres of cardiomyocytes. The hiPSC-CM demonstrated longitudinal alignment in EHTs constructed with and without hiPSC-CF (Figure 7). Cardiac fibroblasts were present in EHTs constructed with hiPSC-CF and were absent in the hiPSC-CM only tissues (Figure 8).
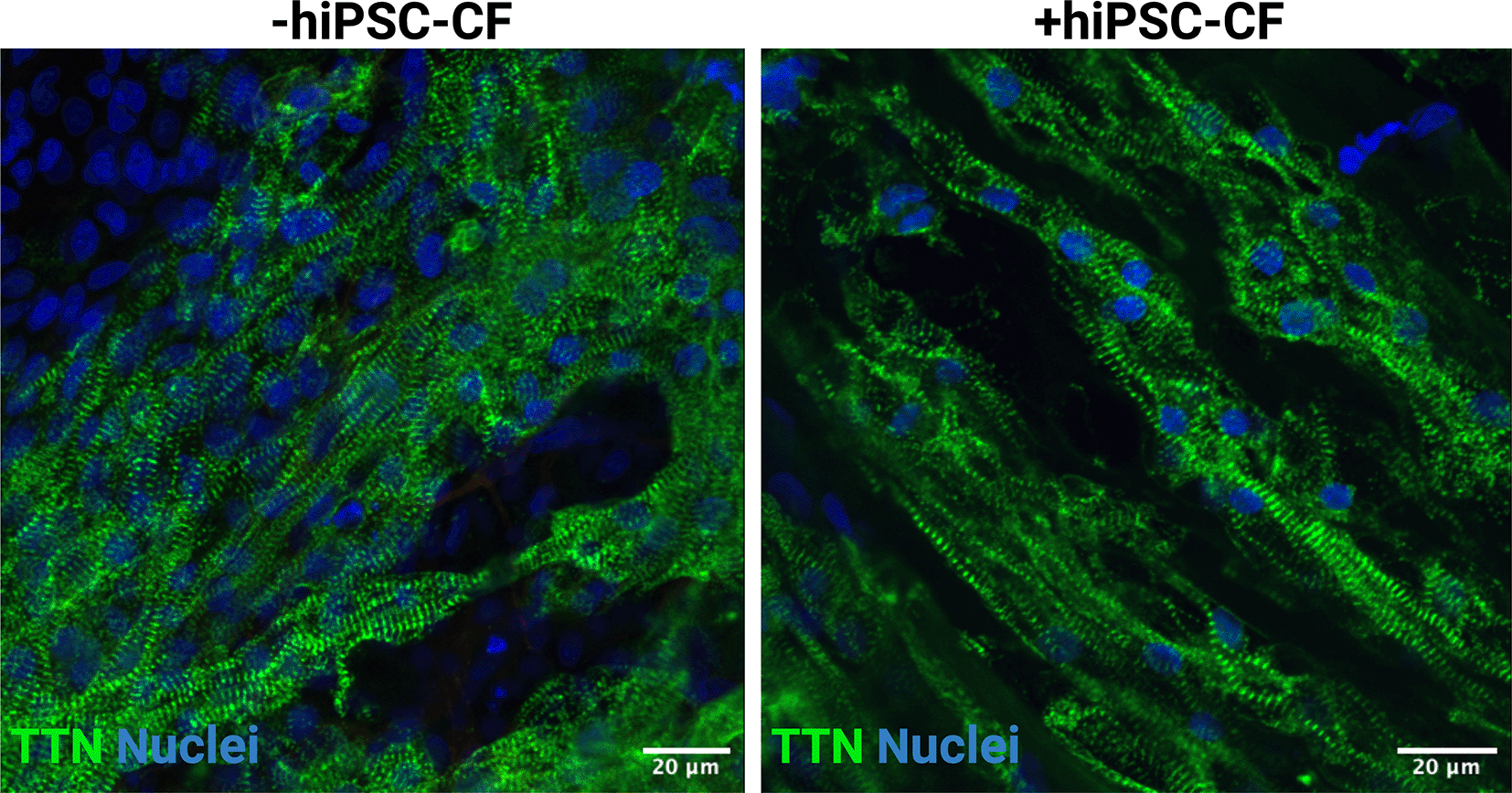
EHTs were constructed with and without iPSC-CFs. Tissues were fixed, sectioned and stained for titin (TTN) and DAPI. Scale bar = 20 microns.
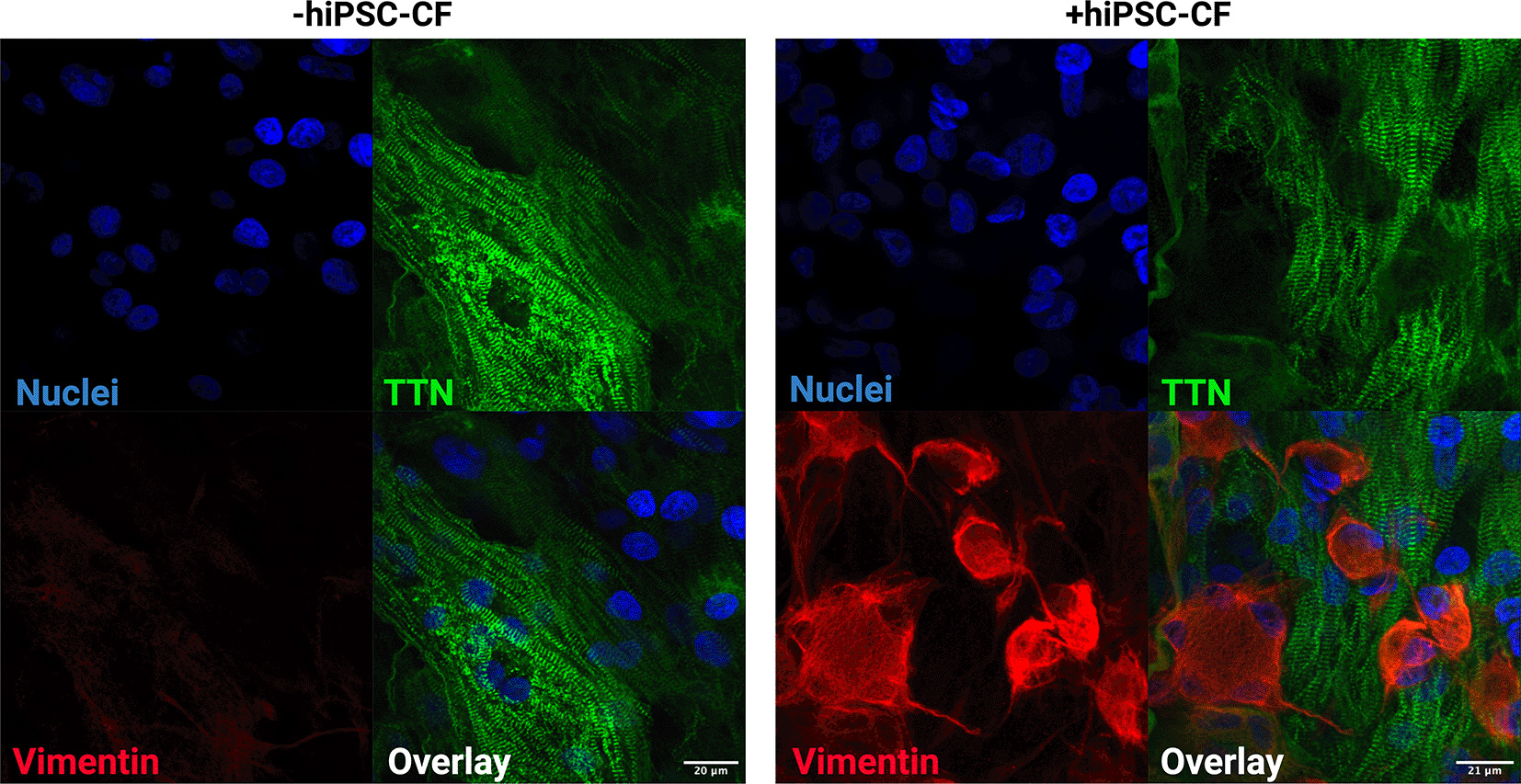
EHTs were constructed with and without iPSC-CFs. Tissues were fixed, sectioned and stained for titin (TTN), vimentin and DAPI. Scale bar = 20 microns.
In this study, we described efficient and reproducible methods for the generation of hiPSC-CM and hiPSC-CF. Furthermore, we described the subsequent integration of the cells into co-culture EHTs with improved contractile function. This approach has potential to explore the complex interplay between cardiomyocyte and cardiac fibroblast signalling involved in pathophysiology. The hiPSC-CF generated were quiescent under the culture conditions outlined in the protocol, with plasticity demonstrated through the addition of the pro-fibrotic cytokine TGF-β1. EHTs constructed with the addition of hiPSC-CF exhibited anisotropic alignment, decreased beat rate, increased tissue compaction and enhanced contractile force. For the first time, we provide comprehensive methods for the generation and addition of quiescent hiPSC-CFs into EHT models. With the resulting model differing from previously described models by the origin of the cardiac fibroblasts (hiPSC derived) (Rivera-Arbeláez et al., 2022) and the type of 3D cardiac model the cells are incorporated into EHT (Giacomelli et al., 2020; Thomas et al., 2021).
The importance of non-myocyte cell types in 3D cardiac constructs is well established and has been widely discussed in previous publications (Kofron and Mende, 2017). EHTs consisting of both hiPSC-CM and hiPSC-CF demonstrated an increase in tissue compaction following just 72 hours in culture and a decrease in beat rate after two weeks in culture. Tissue compaction plays an important role in determining contractile capability and likely occurred in the EHTs constructed with hiPSC-CF due to the regulatory function that cardiac fibroblasts have on extracellular matrix (Rivera-Arbeláez et al., 2022; Thavandiran et al., 2013). A decrease in beat rate was observed from EHTs that were constructed with hiPSC-CF. This is consistent with previous studies that have demonstrated lower beating rate frequencies in hiPSC-CM/fibroblast co-culture models and is likely due to increased CM maturity and decreased funny current expression (Ronaldson-Bouchard et al., 2018).
EHTs constructed in this study from metabolically purified populations of hiPSC-CM, without the addition of hiPSC-CF, demonstrated diminished or no contractile function. Functional and contractile EHTs consisting solely of hiPSC-CM were generated during the course of this study but with a significantly reduced rate of success. The presence of non-myocyte cell types in the tissue population has previously shown to be beneficial to the successful generation and improved contractility of 3D cardiac models (Naito et al., 2006; Ravenscroft et al., 2016). Some studies have robustly demonstrated the successful generation of EHTs using CM populations with a purity of 92-100 % (Mannhardt et al., 2016, 2020). Differences in stem cell line, hiPSC-CM differentiation, handling, and maturation may help explain contrasting success rates of hiPSC-CM only EHT construction. The further development of 3D cardiac models consisting of defined populations of cardiac myocytes and stromal cells, e.g., fibroblasts, will help to reduce batch-to-batch variability and contribute to the development of more reliable in vitro modelling systems.
The relative ease by which hiPSCs can be genetically engineered has allowed researchers to efficiently interrogate the effects pathogenic variants have in the cardiac myocyte. The development of isogenic EHTs consisting of hiPSC-derived cardiomyocytes and cardiac fibroblasts provides a more physiological model in which mutations can be explored and presents increased opportunities for exploring variants with pathological effect in both cell types (Zou et al., 2022). Furthermore, such models represent an exciting prospect for the future incorporation into pre-clinical drug screening and may eventually help relieve the current reliance on in vivo models.
Differences in the intrinsic electrophysiological properties of mouse cardiac myocytes have hampered efforts to model complex cardiovascular diseases. Furthering our understanding of currently enigmatic aspects of human cardiac disease, such as the dynamic interplay between cardiac fibrosis and electrical remodelling, likely requires physiological and ultimately human models, such as the EHTs applied in this study. Improvements to human in vitro modelling systems are required to understand the complexities of cardiac pathology and will likely aid in the reduction and/or replacement of animal models in cardiac research.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)