Keywords
nicastrin, docking, molecular dynamic simulations, binding site, free energy of binding, druggability, breast cancer
This article is included in the Cell & Molecular Biology gateway.
nicastrin, docking, molecular dynamic simulations, binding site, free energy of binding, druggability, breast cancer
Breast cancer's devastating effects are revealed by high incidence and mortality rates worldwide. Breast cancer is the most commonly diagnosed cancer, accounting for 11.6% of all cases and being the leading cause of cancer deaths in women, according to GLOBOCAN estimates from 2018.1–3 Because the estrogen receptor alpha (ER) stimulates more than 75% of breast cancer cases, most standard breast cancer therapies inhibit the function of ERα.4,5 However, the success is obscured by 40 to 50% resistance due to alternative signaling pathways that fuel the growth of breast cancer cells6,7 and overexpression of nicastrin, a member of a four-contained gamma-secretase complex implicated in the resistance mechanism.6,8,9 The resistance mechanism relies on breast cancer stem cells, a subpopulation of cancer cells that drive breast cancer progression and recurrence, and these cancer stem cells thrive on signaling pathways critical to normal stem cell survival, for example, the notch signaling pathway. The notch signaling pathway is processed by the gamma-secretase complex, which results in the formation of the notch intracellular domain (NICD), which when released into the nucleus initiates a variety of cellular processes such as stem cell maintenance, cell differentiation, and cell death.6 The notch-signaling pathway must be modulated for standard breast cancer drugs to be effective. As shown in Figure 1, the gamma-secretase complex is composed of presenilin, presenilin enhancer-2 (pen-2), anterior pharynx defective-1 (aph-1), and nicastrin.
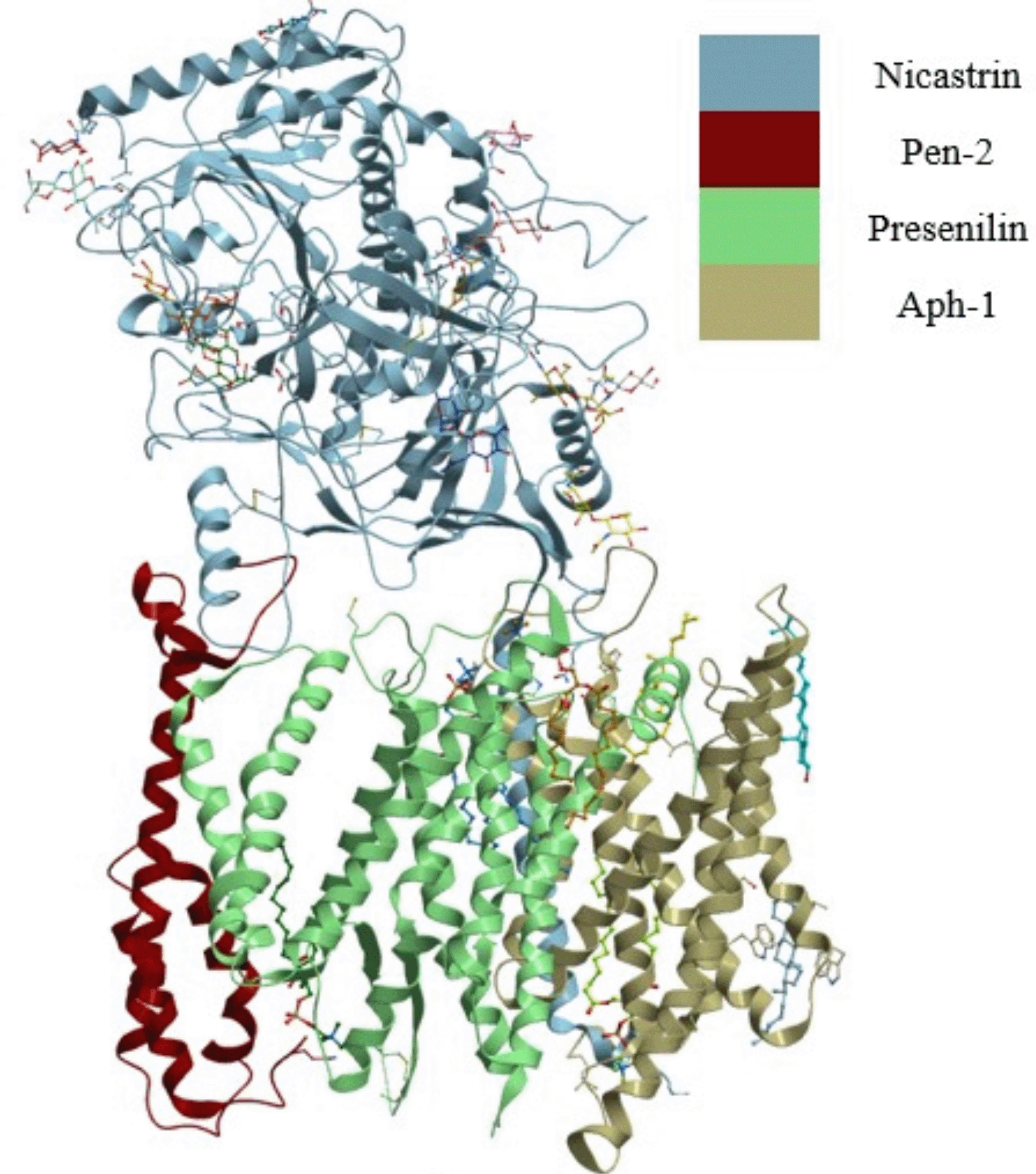
The four subunits are shown as presenilin, pen-2, aph-1, and nicastrin.
Presenilin bears the catalytic site of the complex and pen-2 directly binds to presenilin and contributes to the maturity and catalytic mechanism of the complex.10 Aph-1 and nicastrin function to stabilize the complex11 and nicastrin are also involved in substrate recognition and recruitment.12,13 The modulation of the notch can be done by targeting the gamma-secretase complex that mediates its proteolysis, however, inhibition of the gamma-secretase complex by targeting the catalytic site has confounding effects due to its functional link to critical signaling processes. Various studies have looked at modulation of the gamma-secretase complex rather than its inhibition to avoid these confounding effects on normal cell processes. Since nicastrin is involved in substrate recognition and recruitment and not in the catalytic events of the complex, targeting nicastrin could modulate the functions of the complex without completely inhibiting it.14
Nicastrin is a single-pass transmembrane protein with a heavily glycosylated ectodomain. Functional domains have been identified in nicastrin and are a possible location of binding sites that can be used in designing small molecules that target nicastrin. The ectodomain shares structural similarity with aminopeptidases, especially with the bacterial aminopeptidase (BAP) both having a large and small lobe in the ectodomain. The active site in BAP is located in the large lobe and bears two zinc ions which are essential for protease activity, whilst a similar region in nicastrin is covered by a loop that acts as a lid extending from the small lobe to the large lobe.13 The area covered by the lid in nicastrin does not have the zinc ions for protease activity but bears the conserved hydrophilic DYIGS motif corresponding to residues Asp336, Tyr337, Ile338, Gly339, and Ser340, that are thought to be essential in modulating gamma-secretase activity, and in substrate recognition and recruitment.15,16 The tetratricopeptide repeat-like domain is another functional domain that has been identified in nicastrin. The tetratricopeptide repeat-like region is homologous to the tetratricopeptide repeat domain that is usually involved in peptide recognition. A study by Zhang et al. (2012) revealed that binding an antibody to a conformational epitope in the tetratricopeptide repeat-like domain resulted in the loss of enzymatic activity by the gamma-secretase complex.17 Efforts in targeting nicastrin have seen the development of monoclonal antibodies to modulate the substrate binding and these monoclonal antibodies managed to reduce the production of the NICD.14,18 Recently, it was reported that a notch inhibitor cowanin was found to reduce levels of nicastrin without affecting the expression of other gamma-secretase subunits.19 A search through bioassay data in PubChem20 provides more than 500 small molecules that target nicastrin and other components of the gamma-secretase complex though binding data does not show small molecules that are specific to nicastrin only. Insight into the binding modes and structural basis of interactions of small molecules in nicastrin and the preferred binding sites of these compounds in nicastrin are still to be published.
In silico approaches such as molecular docking can be used to identify binding sites and determine binding modes of small molecules in protein structures. Molecular dynamics can be used to validate the binding site and interactions. An assessment can also be done to ascertain if drug-like molecules can bind to the binding sites. These drug-like molecules possess properties such as solubility predicted by the octanol-water partition coefficient less than 5; molecular weight between 200 and 500 Daltons; polar surface area and charge described by less than 10 H-bond acceptors and 5 H-bond donors.21 These properties influence in vivo and in vitro activity of orally active compounds.
The present work aimed to identify possible binding sites in nicastrin and determine modes of binding of a set of previously reported nicastrin inhibitors. Blind docking calculations were used to predict binding sites in nicastrin. The docking calculations were validated by molecular dynamics simulations to establish the stability of the ligand in the binding site. Binding modes of known nicastrin inhibitors are also reported. The results of this study can be used to enhance the structure-based drug design efforts of anticancer drugs targeting nicastrin.
Protein preparation
The structures of the gamma-secretase complex (PDB ID 6IDF)22 and (PDB ID 5A63)23 were retrieved from the Protein Data Bank and nicastrin (Chain A) was extracted from these structures. Two nicastrin structures were used to select the best conformer during binding mode analysis. Using AutoDock Tools,24 nicastrin structures were prepared for docking calculations by adding Gasteiger charges, merging non-polar hydrogens, as well as assigning the correct AutoDock4 atom types, and adding hydrogen atoms. The prepared Nicastrin structures were saved in pdbqt format.
Ligand preparation
A dataset was obtained from the PubChem database,20 which contained 536 human nicastrin inhibitors with their biological activities expressed as IC50 values. Inhibitors labelled Inconclusive and Unspecified impair the ability of derived models to predict bioactivity and, as a result, were removed from the dataset. Because the data was collected from various bioassay types, the bioactivities were normalised by converting them to molar units and then logarithmically transforming them to pIC50 (-logIC50) values. The data set's inhibitory potencies, expressed as pIC50, ranged from 4.3 to 11.7. With a pIC50 of 8.0, the molecules were classified as active. The dataset was clustered using hierarchical clustering in DataWarrior, and 30 compounds (Figure 2) from the clusters were chosen for docking.
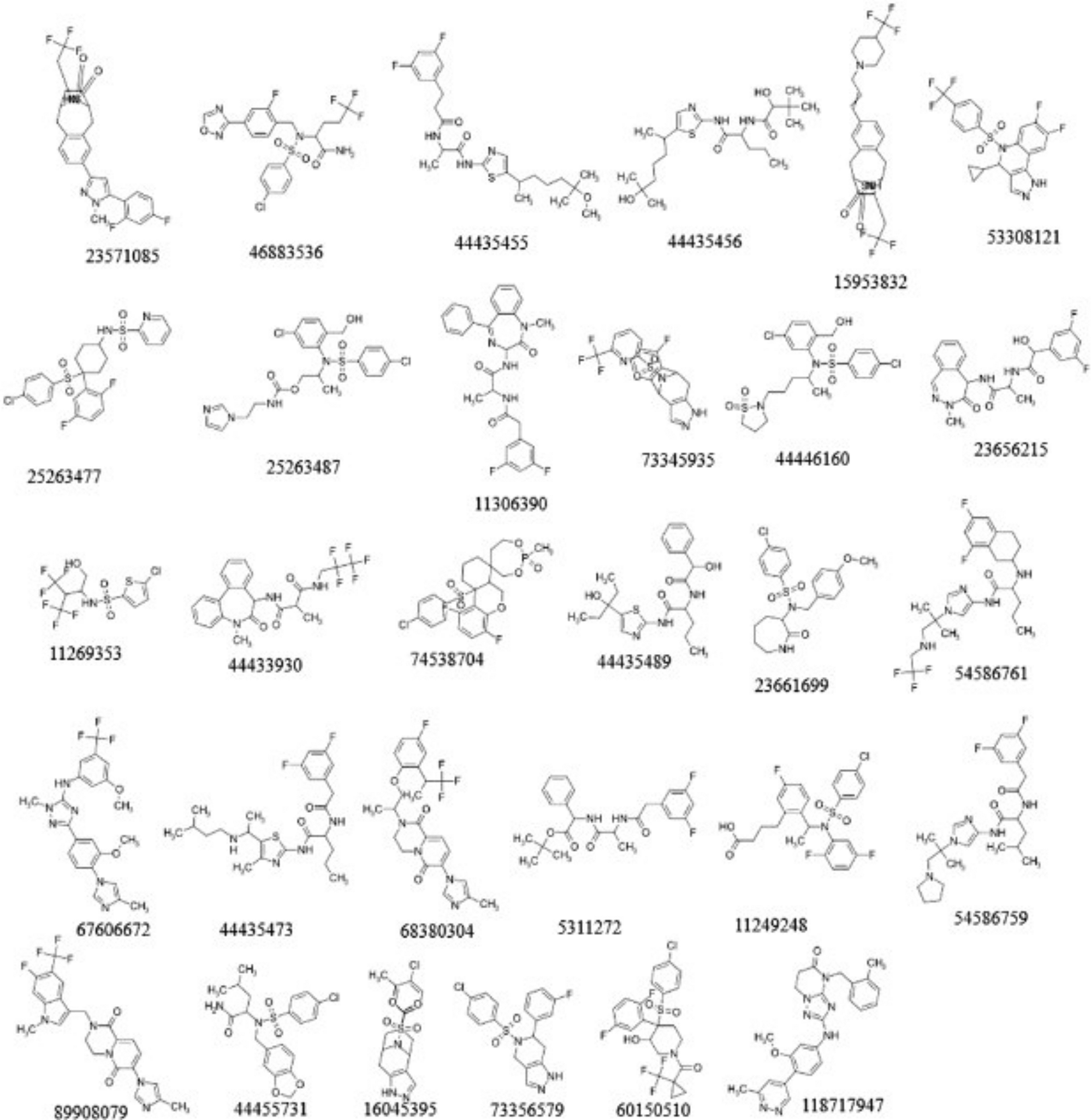
Using AutoDockTools,24,25 the 30 ligands were prepared for docking by carrying out energy minimization for 200 steps using conjugate gradient and MMFF94 force field, adding Gasteiger charges, merging non-polar hydrogen atoms, assigning AutoDock4 atom types, and adding hydrogen atoms. The root, torsion degree of freedom, and the number of rotatable bonds were defined for each of the ligands. The ligand structures were saved in pdbqt format.
Blind docking in Autodock Vina was done to identify potential binding sites in nicastrin. From the docking set of 30 ligands, ligand CID 44433923 ([1-[[(7S)-5-methyl-6-oxo-7H-benzo [d][1]benzazepin-7-yl]amino]-1-oxopropan-2-yl]N-(2,2,3,3,3 pentafluoropropyl)carbamate) with a high nicastrin bioactivity from PubChem was selected for use in the blind docking calculations. The grid box for docking was centered on the protein with a 0.375 Å default spacing and x,y,z dimensions of 70.6636 × 122.1096 × 64.2142 Å and 92.3445 × 67.4301 × 108.9436 Å for 5A63 and 6IDF, respectively.
The Internal Coordinate Mechanism (ICM) method developed by Molsoft L.L.C26 was used to validate the predicted binding sites of the two protein structures (PDB IDs 6IDF and 5A63). Using receptor preparation tools in ICM, the protein structures were individually prepared by optimizing hydrogen, histidine, proline, glycine, and cysteine residues. Missing hydrogens and heavy atoms were added and the structures were saved as ICM objects. Potential binding pockets on the proteins were identified using ICM PocketFinder27 and their druggability was given by the calculated DLID score.28
To determine the stability of the docked complex as well as the intermolecular interactions over time, molecular dynamics simulations were performed using GROningen MAchine for Chemical Simulations (GROMACS) 2022.129 software and Chemistry at Harvard Macromolecular Mechanics (CHARMM36)30 as an all atom forcefield. The AnteChamber Python Parser interface (ACPYPE)31 portal was used to generate ligand topology for CID44433923. The complex was solvated in an octahedral TIP3P water-box with a distance of 10 Å between the box's edges and neutralized using K+ and Cl- ions. This was followed by a steepest descent method used to minimize the system and set at 5000 steps. The steepest descents converged at 2368 steps when the maximum force was less than 1000 kJ/mol/nm. The system was equilibrated for 125 ps and all bonds and heavy atoms were restricted by the LINCS (Linear Constraints Solver) algorithm. The temperature and pressure were set to 310 K and 1 atm respectively and finally the system was subjected to a 50 ns production run saving the trajectories every 2 ps.
To determine the stability of the docked complex as well as determine the intermolecular interactions over time, the root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (RoG) and number of hydrogen bonds (hBond) were obtained from gromacs routines for the analysis.
Free energy of binding was calculated using molecular mechanics with generalized Born surface area solvation (MMPBSA) using gmx-mmpbsa.32 These calculations considered snapshots from 27 to 50 ns of the molecular dynamic simulations. The binding energy calculated considered only the enthalpy of a single trajectory to minimize computing costs. Per residue energy decomposition analysis was also done to identify residues in the binding site contributing to the binding energy.
To characterize the binding modes and interactions of known nicastrin inhibitors, the 30 prepared ligands were docked into the DYIGS binding site of nicastrin PDBID 6IDF using the Autodock Vina tool. The grid box for docking was centered on the protein at 171.8221, 192.4385, 218.7799 Å with a 0.375 Å and x,y,z dimensions of 42.6757 × 41.9425 × 42.4690 Å.
Initially, blind docking calculations was performed to predict potential binding sites in nicastrin conformers. The blind docking predicted three distinct binding sites which are located in similar locations in both conformers. The identified sites (Table 1) encompass domains or signature regions within nicastrin that are unique to their function (Figure 3A).33 These include a site that contains the DYIGS signature34 (DYIGS site) and the Tetratricopeptide region like site17 including a potential binding site positioned in a central cleft in the hinge region (Hinge region site). The DYIGS site had a higher affinity for compound CID44433923 as compared to the two other sites.
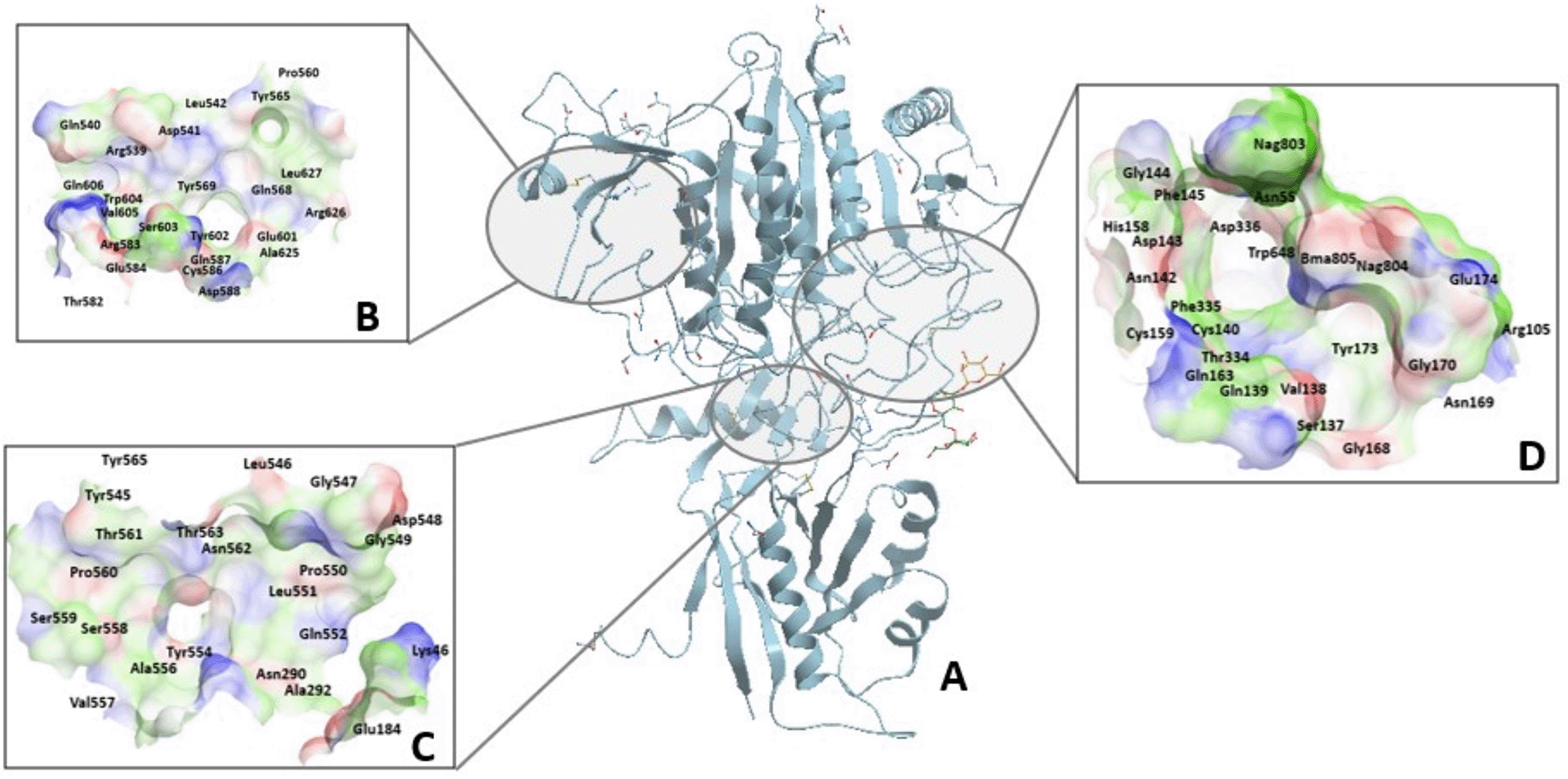
A) The binding sites are situated and hence named according to functional regions in nicastrin. These are shown as B) Tetratricopeptide repeat-like site, C) Hinge site and D) DYIGS site all located in the large lobe of the protein.
A shallow pocket located on the surface of the large lobe was predicted as a binding site and contains the tetratricopeptide repeat-like domain (Figure 3B). The tetratricopeptide repeat-like domain is homologous to TPR domain 2A and 2B helices of the HOP human protein, which binds to the C-terminal peptide of Hsp90. The TPR domains bind to substrates via side chains of α-helix residues.
A hinge region that is conserved in nicastrin homologues facilitates the rotation of the large lobe relative to the small lobe13 during substrate binding.12 The hinge region is composed of phenylalanine residues (Phe286 and Phe287) from the large lobe which interact with side chains of phenylalanine residues from the small lobe through van der Waals interactions. This region forms a central cleft35 that is located at the back of the DYIGS pocket. Blind docking studies identified the central cleft and the residues that surround it as a potential binding site. Figure 3C shows the hinge site.
The DYIGS site (Figure 3D) houses the DYIGS motif corresponding to residues Asp336, Tyr337, Iso338, Gly339, and Ser340 are situated in the large lobe.12,16,36 These hydrophilic DYIGS residues are proposed to bind to hydrophilic N-termini of substrates.13 The hydrophilic DYIGS residues are buried under a hydrophobic loop or lid that extends from the small lobe.37 The orientation of the hydrophobic loop residues exposes just the side chains of Asp336 oriented towards the surface for interactions. During docking, ligands interact with the lid residues that line the hydrophilic pocket rather than interacting with the conserved residues. The hydrophobic environment conferred by the lid encourages ligand binding via hydrogen and hydrophobic bonds and ion pair interactions. The presence of aromatic residues such as phenylalanine (Phe145, Phe335, and Phe448), histidine (His58, His158, and His444), tryptophan (Trp648), and tyrosine (Tyr173, Tyr337, and Tyr453) is known to influence the function as well as molecular recognition in proteins and their presence in nicastrin might influence substrate recognition and recruitment.38 Glycans in the binding site also control the accessibility of the binding site.39
The geometry and physicochemical properties of the binding site are important in establishing the ability of the binding site to bind drug-like molecules. Descriptors that characterize geometry include volume, surface area as well as buriedness of the binding site. These descriptors correspond to the shape and size of small molecule binders of that site.27,40,41 Physicochemical properties of the site compliment the drug-like nature of the drug.27 ICM PocketFinder27 was used to assess the geometry and physicochemical properties as well as the druggability of binding sites identified by blind docking studies.
The three distinct sites identified through blind docking studies and a summary of their druggability assessment are provided in Table 2. Two of these sites (DYIGS site and Hinge site) had DLID scores favorable for binding drug-like molecules. The differences in the geometric features of these sites in the conformers might be explained by ligand-induced changes in nicastrin that were observed by Bolduc et al. (2016).12 When notch binds to the transmembrane domain of the gamma-secretase complex, rotation of the hinge region (residue Phe287) in nicastrin is induced which affects the hydrogen bond network and flexibility of the ectodomain,12 resulting in increased volume of the binding sites. The increased volume in PDB ID 6IDF can positively impact the free energy of binding ligands within the binding site.
The Hinge site in both conformers has the same hydrophobicity and approximately the same buriedness, however, the 6IDF conformer has higher aromaticity and volume and hence a higher DLID score. The volume of the 6IDF conformer is approximately 200 Å3 (Table 2) and volumes of most ligands in the dataset average 250 Å3. Considering that the volume of the ligand is known to be correlated to that of the binding site, with the ligand rarely occupying the entire binding site, binding modes of ligands were not assessed in the hinge site due to the small volume of the binding site relative to the volume if the ligands in the data set.
The tetratricopeptide repeat-like site, though having the second-largest volume had negative DLID scores in both conformers due to its low hydrophobic and aromatic character. The negative DLID score shows preferential binding to highly polar molecules that are not drug-like.
An assessment of the druggability landscape of nicastrin (PDB ID 6IDF) was done since it was the most druggable conformer (Table 2). The assessment was done by comparing attributes that define a druggable binding site using a druggability landscape. The attributes include volume, hydrophobicity, and buriedness. In the druggability landscape (Figure 4), volume was plotted against hydrophobicity and coloured according to buriedness, with larger dots indicating a druggable site. The druggability landscape shows the twenty pockets identified in ICM PocketFinder, which include the DYIGS, tetratricopeptide repeat-like, and hinge sites predicted through blind docking. ICM PocketFinder provides a DLID score which is a druggability assessment to ascertain the potential of the binding sites to interact with drug-like compounds. A positive DLID score close to 1 assumes a site to be targeted by drug-like molecules.28 Out of the twenty binding sites that were identified, just two (DYIGS site and hinge site) were predicted to be druggable by drug-like molecules.
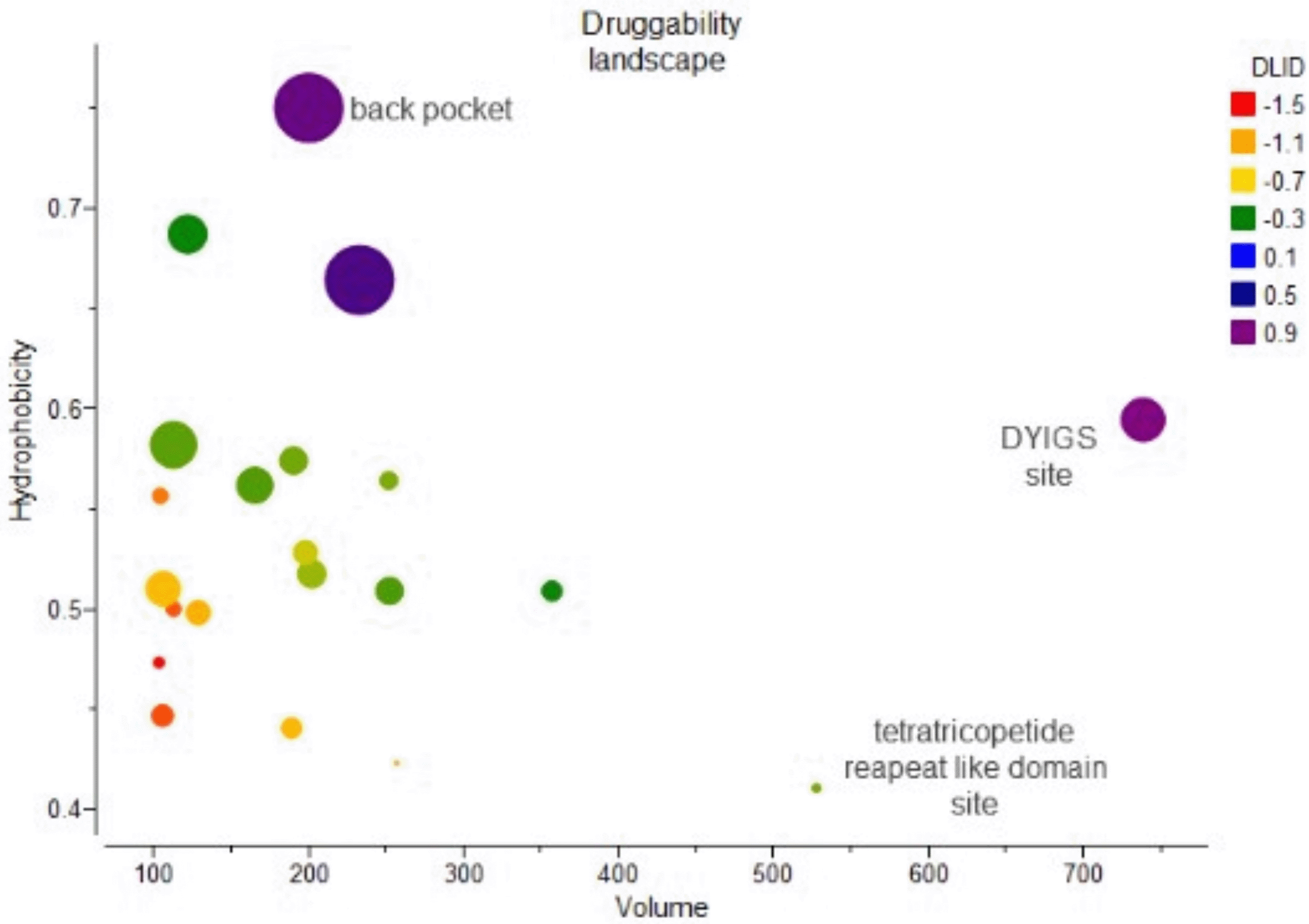
Predicted binding sites are represented by dots coloured according to their DLID scores with a positive high DLID score (purple) denoting a very druggable site and a negative DLID (red) score describing a site that is very difficult to target using drug-like molecules.
The landscape shows that the druggability of binding sites in nicastrin increases with hydrophobicity. This is understandable as hydrophobicity dominates free energy binding in protein-ligand interactions.42 Considering the characteristics of the DYIGS site which is covered by a hydrophobic lid and the hinge site which is surrounded by hydrophobic residues like Phe103, Leu171, Phe176, and Ile180, this greatly contributes to their hydrophobicity.13
To determine the stability of the docked complex and understand intermolecular interactions over time, molecular dynamics simulations were carried out. To achieve this, the root mean square deviation (RMSD), root mean square fluctuation (RMSF) and radius of gyration (RoG) was obtained from gromacs routines for the analysis.
The RMSD describes the overall conformational stability of the protein-ligand system by calculating the changes in the protein's carbon alpha (Calpha) and primary conformation, as well as that of the ligand's, during the simulation timescale. The results show that there were no major conformational changes in nicastrin as indicated by the RMSD of the complex (Figure 5). This suggested that within the 50 ns simulation the ligand was stable within the DYIGS binding site.
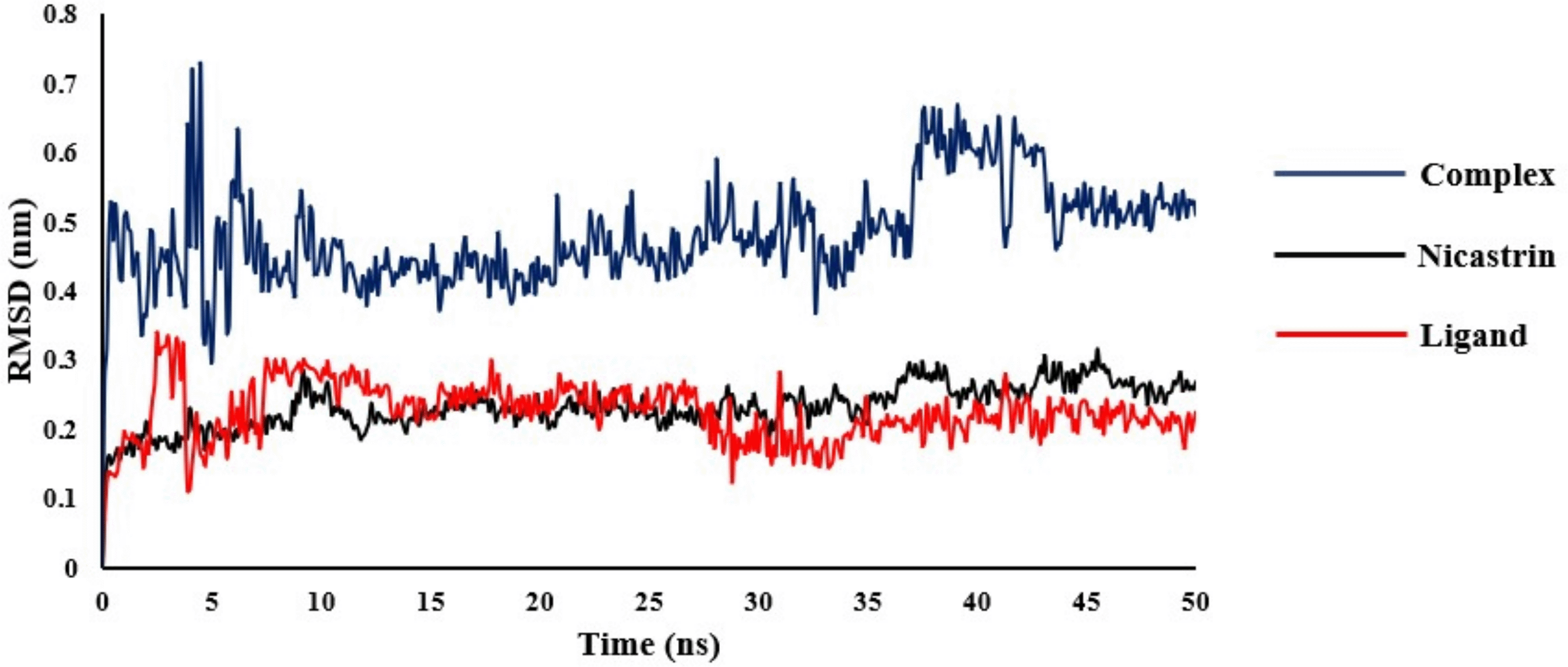
The RMSD for nicastrin was found in the range of 0.11-0.32 nm with an average of 0.23 nm and from 0.07-0.34 nm with an average of 0.22 nm for the ligand. The complex of nicastrin bound to CID44433923 had an RMSD in the range 0.27-0.72 nm with an average of 0.48 nm.
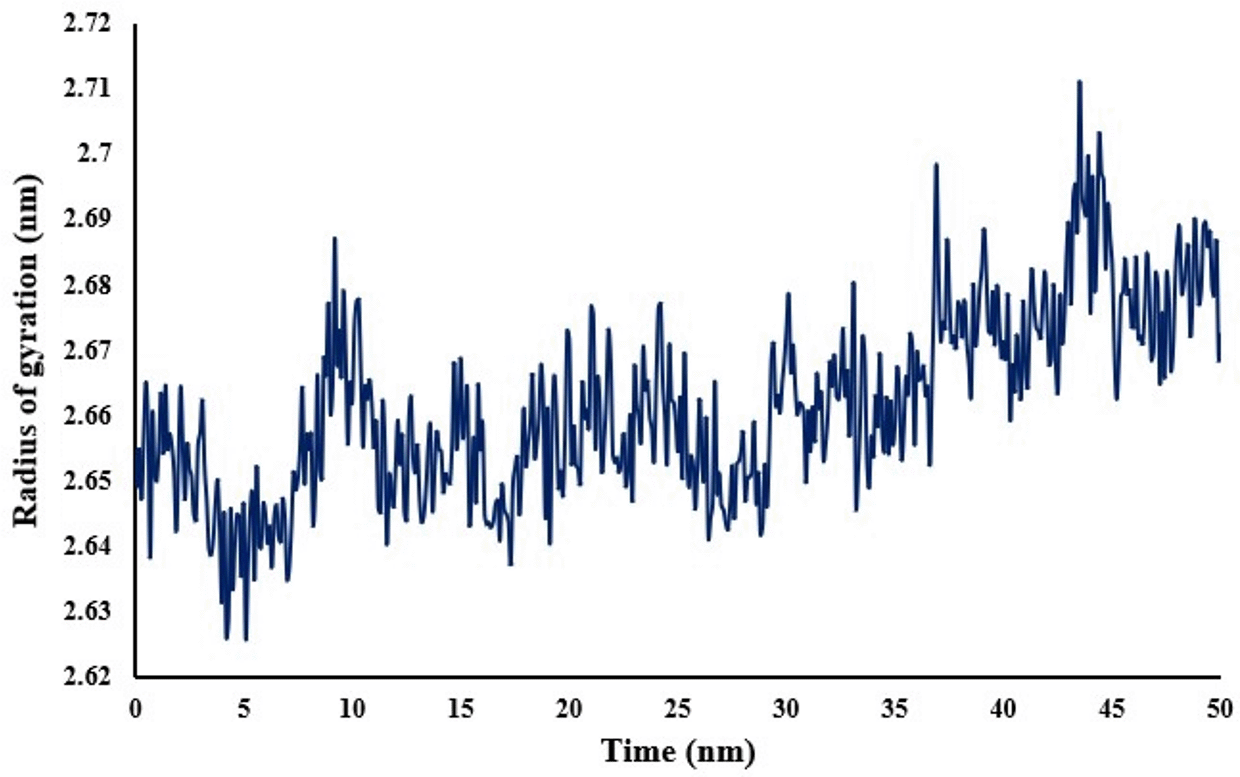
To account for the structural integrity of nicastrin when bound to the ligand, the RMSF (Figure 6) was evaluated. The lower the RMSF values per amino acid residue, the more stable, rigid and compact the receptor. A low RMSF value of binding site residues of not more than 0.54 nm shows that the ligand was stable within the binding site. The compactness and stability of nicastrin is also shown by its radius of gyration in Figure 7. The complex is compact as shown by the range of radius of gyration of 2.65-2.67 nm with an average of 2.66 nm.
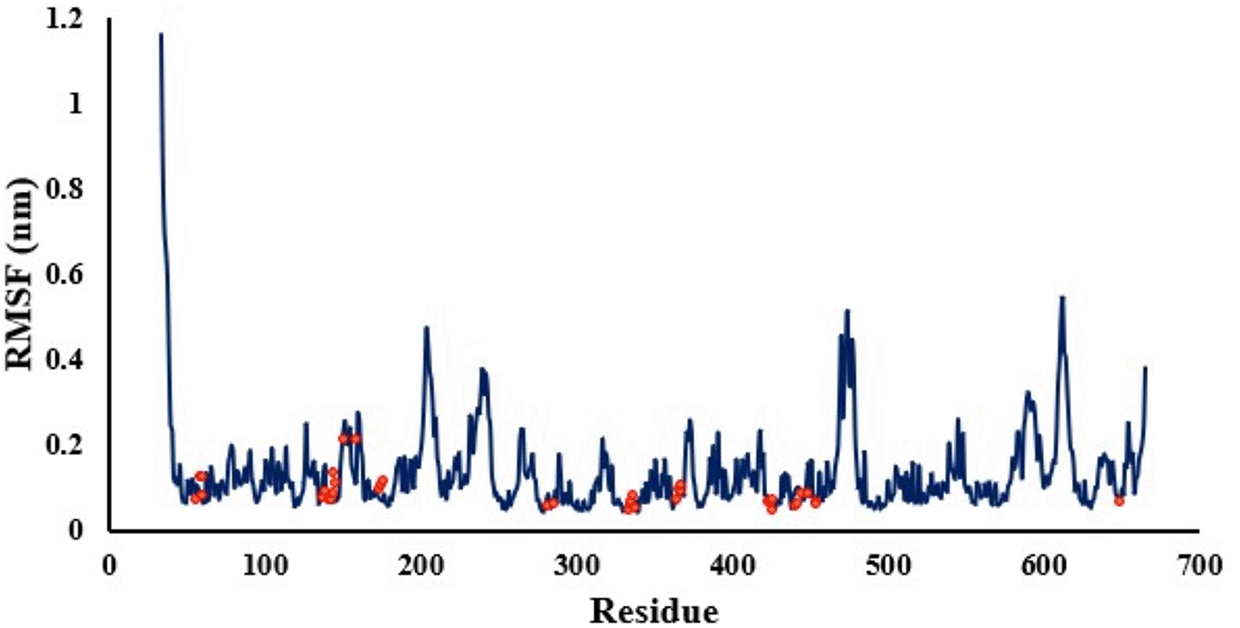
The red dots indicate binding site residues identified.
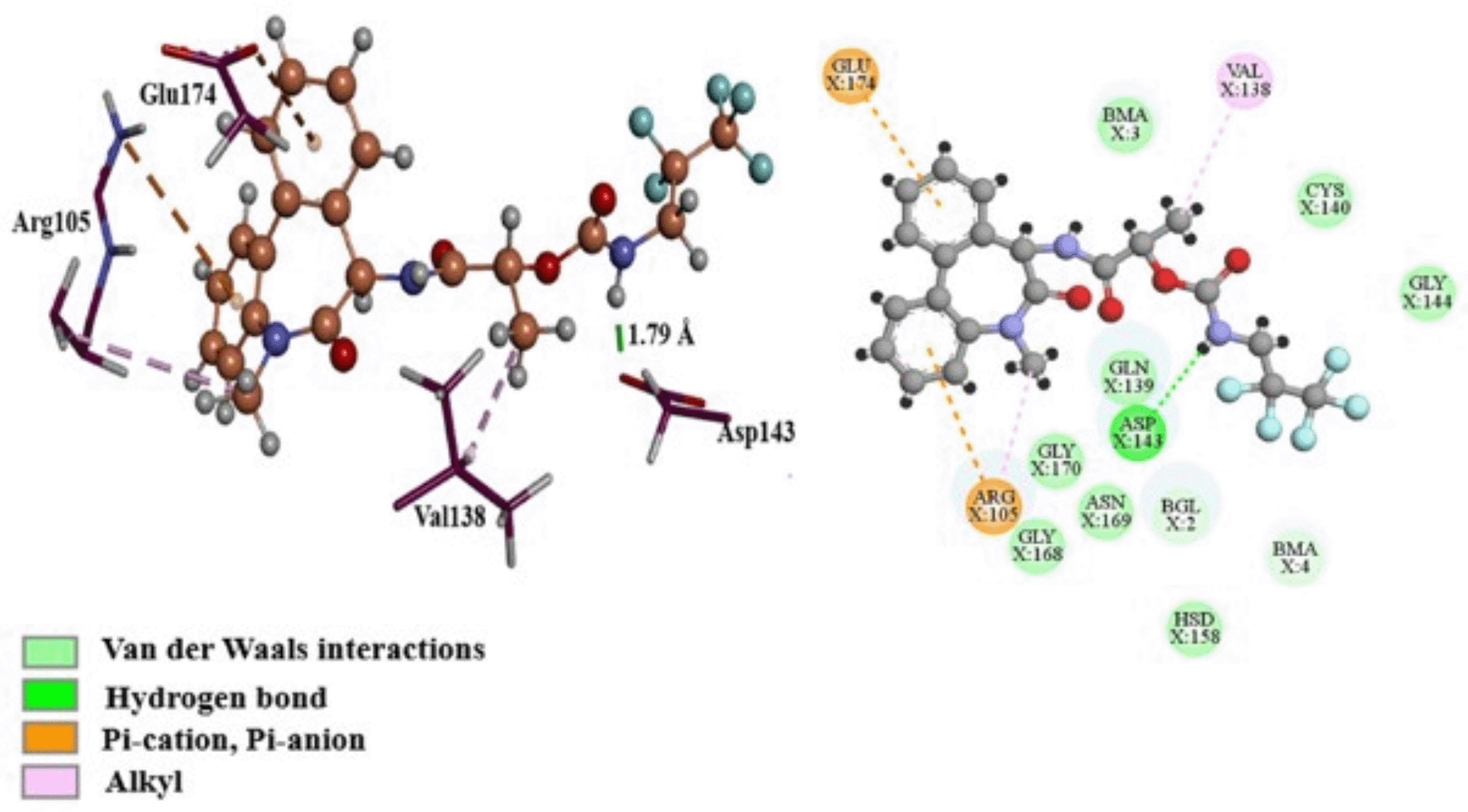
The Gibbs free energy of -11.40 kcal/mol for ligand binding to nicastrin was calculated using MM/GBSA, as indicated in the Table 3. The van der Waals energy contribution was -21.24 kcal/mol and since this was the lowest energy term it shows that binding free energy is mainly driven by hydrophobic interactions. On the other hand, the electrostatic energy was -15.15 kcal/mol which shows its importance in the binding of ligands to nicastrin in the DYIGS site. To better understand the interactions that affect the binding's free energy the conformation with the lowest binding energy was examined. This conformation was found at 45.6 ns with a binding energy of -19.34 kcal/mol and is presented in Figure 8. The residues Gln139, Cys140, Gly144, His158, Gly168, Asn169, Gly170, and the glycan Bma805 were involved in van der Waals interactions. Along with Val138, Arg105 had a role in hydrophobic alkyl interactions. Also, the presence of Arg105 and Glu174 encouraged electrostatic interactions, with the positively charged Arg105 creating pi-cation contacts and the negatively charged Glu174 creating pi-anion interactions with the aromatic rings. Asp143 created a strong hydrogen bond of 1.79 Å by donating a hydrogen from the amide hydrogen. Per residue free energy decomposition using MM/GBSA identified residues Gln139, Val138 and Arg105 as contributing to the binding energy the most, which indicates their importance in nicastrin activity. These residues can be used in selecting small molecules during drug design.
| Energy component | VDWaals | EEL | EGB | ESURF | GGAS | GSOLV | TOTAL |
|---|---|---|---|---|---|---|---|
| Δ Energy | -21.24 | -15.15 | 27.96 | -2.96 | -36.39 | 24.99 | -11.40 |
To analyse binding modes of known nicastrin inhibitors, the selected 30 ligands (Figure 2) were docked into the DYIGS site. These compounds in the data set were first grouped according to their common substructures. A total of eight groups were obtained and the common substructures are shown in Figure 9. Groups 1, 2, and 3 contain the sulfonamide group, with groups 2 and 3 having the sulfonamide group linked to a phenyl ring. Groups 4, 5, and 6 are related through the 3-aminopropanamide component within their maximum common substructures. Generally, the analysis shows that these compounds interact with residues Val138 and Gln139 which were identified through per residue free energy decomposition analysis as contributing to the binding energy in the DYIGS site in nicastrin.
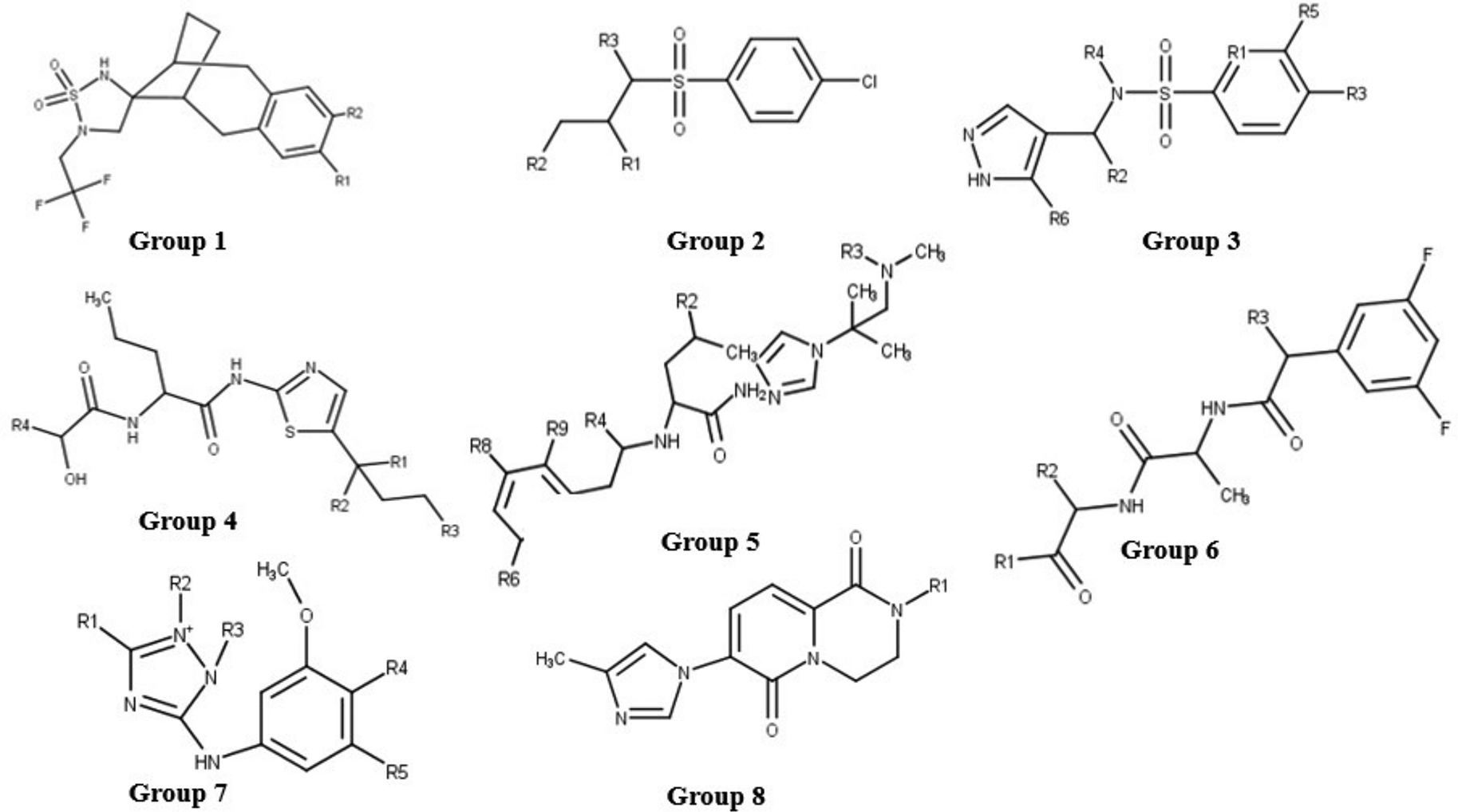
Compounds that bear the sulfonamide group are generally known to be effective with good pharmacokinetic properties.43 This is observed in the diverse set used in this study, sulfonamide bearing compounds are more potent (IC50 values ranging between 6.0×10-5 μM and 8.3×10-3 μM) than other compounds without the sulfonamide group. Group 1 consists of a spiro[1,2,5-thiadiazolidene-4,13’-tricyclo[8.2.1.03,8]trideca-3(8),4,6-triene] 1,1-dioxide substructure with two compounds; CID 23571085 and CID 15953832 bearing this moiety. These compounds are oriented similarly in the binding site; with the spirocyclic sulfonamide group predicted to interact with Val138 and Gln139 through hydrophobic interactions. Hydrogen bonding was observed between residues Tyr173 and trifluoromethyl and difluorophenyl groups in CID 15953832 and CID 23571085 respectively. Group 2 has eight compounds and contains the 4-chlorobenzene sulfonamide moiety. The sulfonyl component mostly interacts with Val138 and Gln139 through hydrophobic interactions just as with Group 1 compounds, however salt bridges between Asp 143 and Asp336 and the tert-amine group on the sulfonamide are also observed. The orientation of Group 2 compounds differs from that of Group 1 in that the halogenated phenyl part of the substructure is mostly anchored into the binding site via hydrophobic contacts with the glycans.
Considering Group 3 with four compounds containing the methyl pyrazole conjugated to a substituted benzenesulfonamide group (CIDs 53308121, 73356579, 73345935, and 16045395). Fluoro and chloro substituents in the compounds are suggested to interact with Nag803 and Nag804 through hydrophobic interactions, similar to the orientation of Group 2 4-chlorobenzene. However, the orientation of the sulfonyl part of Group 3 interacts with Cys140. This differs from the interaction of sulfonyls in Groups 1 and 2 which interact with Val138 and Gln139. Hydrogen bonds and hydrophobic contacts with Val138, Asp143, His158, and Bma805 were also common with the pyrazole group.
The binding mode of compounds that contain the sulfonyl moiety is elaborated by the schematic representation of the orientation of CID 15953832 and its interactions in the DYIGS binding site (Figure 10A and B). CID 15953832 is a potent gamma-secretase inhibitor.22 Hydrophobic interactions with residues Val138, Gln139, Asn142, Asp143, Cys159, Tyr173, Asp336 and Trp648 were observed. Interactions from docking show hydrophobic contacts between the fluorine of the difluorophenyl part of the inhibitor and the Asp336 residue, which is part of the DYIGS motif. A salt bridge between the tertiary amine group of the methylpyrazole with Asp143 was established. Hydrogen bonds are revealed between Gln163 and oxygen on the sulfonyl centre.
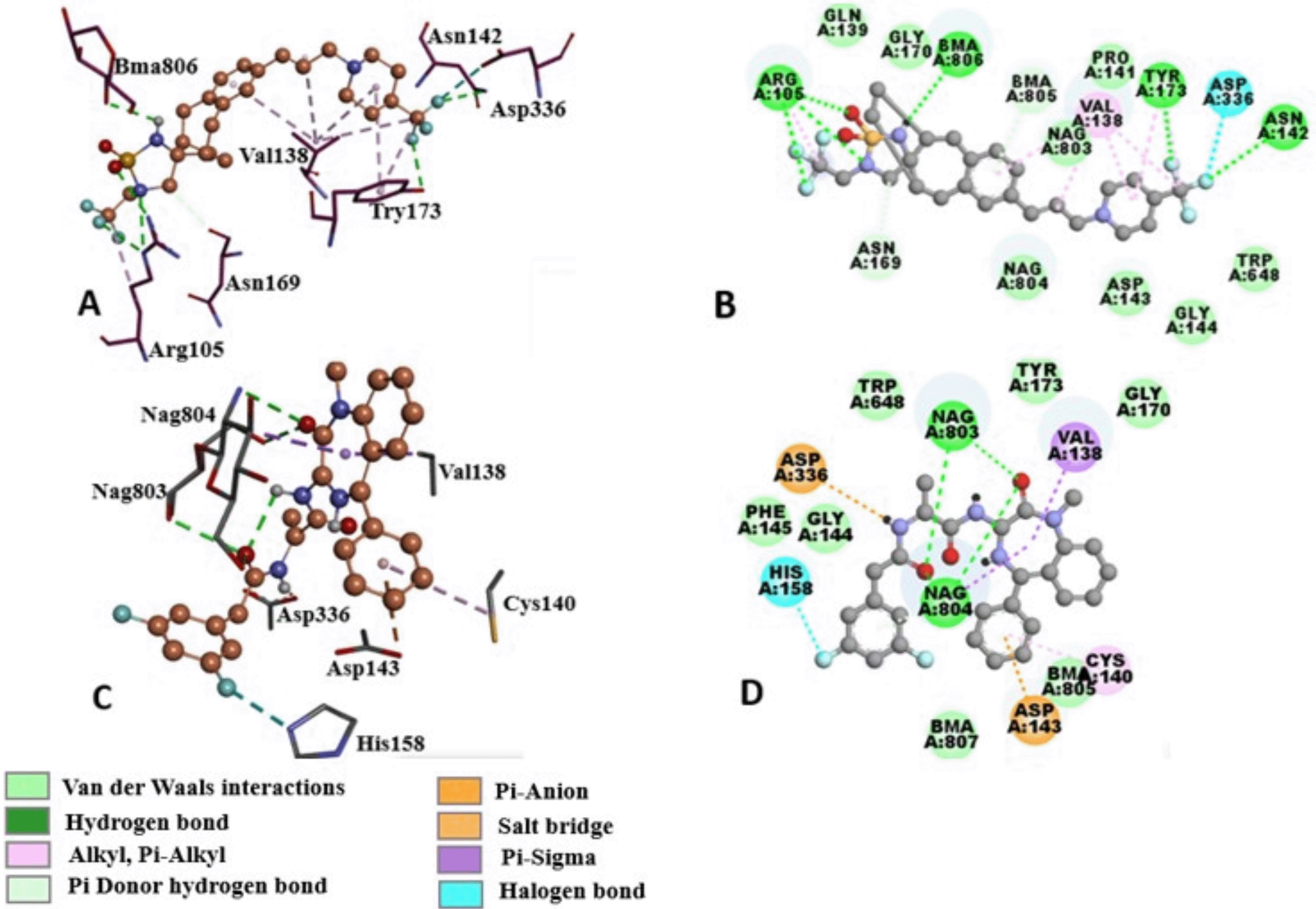
A. Glycans in the periphery of the binding site are depicted as green (NAG) and orange (BMA) balls and sticks. B. 2D representation of binding interactions between compound CID 23571085 and DYIGS binding site residues. Green dashed lines represent hydrogen bonds and the red arcs depict hydrophobic contacts. C. Docked pose of compound CID 11306390 (magenta) in the DYIGS binding site. Glycans in the periphery of the binding site are depicted as green (NAG) and orange (BMA) ball and sticks. D. 2D representation of binding interactions between compound CID 11306390 and DYIGS binding site residues. Green dashed lines represent hydrogen bonds and the red arcs depict hydrophobic contacts.
The 3-aminopropanamide component common in Groups 4, 5, and 6 compounds interacts with residues Val138, Asp143, Nag803 and Nag804 mostly through hydrophobic interactions. The two Group 4 compounds (CIDs: 44435456 and CID44435489) have the 1,3-thiazol-2-yl]amino]-1-oxopentan-2-yl] propanamide group in their structures. The 1,3-thiazole interacts with His158 via hydrophobic contacts and the aminopropanamide with Val138. Asp143 nitrogen forms a hydrogen bond with the oxygen on the dimethylbutanamide in CID 44435456 whilst in CID 44435489, the aspartate oxygen and Gly144 nitrogen form hydrogen bonds with phenylacetylamino oxygens.
Compounds in Group 5 contain the phenylacetyl amino [1-(2-methylpropan-2-yl) imidazol-4-yl] pentanamide maximum common substructure. The phenyl group in the substructure is halogenated and electrostatic interactions between the fluorines and nitrogen of Nag804 are observed. The phenylacetyl amino [1-(2-methylpropan-2-yl) imidazol-4-yl] pentanamide substructure interacts with Asp143, Cys140, Bma807, His158, Bma805 and Bma806 via hydrophobic interactions in both compounds. These compounds also interact with Asp336. The nitrogen on the trifluoromethylamino group forms hydrogen bonds with the aspartate 336 oxygen, whilst the fluorines form hydrogen bonds with Tyr173 and Asn142.
The 3,5-difluorophenyl-acetylamino propanamide substructure is common to the gamma-secretase inhibitor Compound E (CID 11306390) and CID 23656215 making up Group 6. Group 6 compounds are anchored into the binding site by electrostatic interactions between glycans Nag803 and Nag804 and propanamide oxygens. The diazepine group in both compounds interacts with Val138 and whilst residues Asp143, Tyr173, Nag803 and Bma805 form hydrophobic contacts in both compounds.
The binding mode of Groups that contain the 3-aminopropanamide moiety has been illustrated by the schematic representation of the orientation of CID 11306390 in the binding site (Figure 10C and D). This is a potent L-alanine derivative that was developed for the treatment of Alzheimer's disease.44 Halogen bond between His158 and the difluoro substituent was identified. Nag803 and Nag804 form hydrogen bonds with acetylamino propenamide oxygen and oxygen on the diazepine respectively. Aps336 forms a salt bridge with the nitrogen on the acetylamino propenamide. Asp143 forms a pi-anion interaction with the phenyl group. Hydrophobic interactions between the phenyl-2,3-dihydro-1H-benzodiazepine and Val138, Cys140, Trp648, Tyr173, Gly170 and Gly144 and Phe145 residues were also shown.
Group 7 contains two compounds that have the 3-methoxyphenyl-2-methyl-1, 2, 4-triazol-3-amine. CID67606672 contains two methoxyphenyl groups and are positioned such that one methoxyphenyl oxygen interacts with Asn142 via hydrogen bonding whilst the other methoxyphenyl forms hydrophobic contacts with His158, Bma805 and Bma807. The amino group that bridges the methoxyphenyl group and the triazole group is stabilized by hydrogen bonds with Nag803 and Asp336 via hydrophobic contacts. In CID118717947, the methoxyphenyl oxygen shares a hydrogen bond with Gln163 nitrogen.
The eighth group is composed of two compounds, CIDs 89908079 and 68380304 that share the dihydro pyrido[1,2-a]pyrazine-2,3-diol conjugated to a methyl imidazole group. Upon binding a salt bridge is formed between tertiary amines of pyrazine-2, 3-diol in both compounds with Asp143. CID89908079 displays two of these salt bridges with Asp 143 and also with the tertiary amine of the imidazol portion with Asp336. In addition, a T-π stacking exists between the phenyl group in Tyr173 and the pyrido group.
Docking calculations were used to predict binding sites in this study. Three binding sites in nicastrin were discovered through binding site analyses. The druggability of these binding sites was assessed, and the DYIGS site was found to be the most druggable by drug-like compounds. Molecular dynamics simulations, free energy calculations, and residue decomposition analysis were used to validate the binding site. The analysis reveals that hydrophobic interactions and electrostatic forces dominate binding in nicastrin. Residues Arg105, Gln139 and Val138 were identified as the residues contributing the most to the binding energy. Known nicastrin compounds were docked in the DYIGS site and the interactions reveal that they interact with the residues Arg105, Gln139 and Val138. These findings lay the groundwork for developing small molecules targeting nicastrin for anticancer drug discovery.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)