Keywords
Genome annotation, non-model unicellular eukaryotes, gene prediction, evidence data acquisition, Singularity container, automation
This article is included in the Bioinformatics gateway.
Genome annotation, non-model unicellular eukaryotes, gene prediction, evidence data acquisition, Singularity container, automation
Coding sequences (CDS) and, more generally, gene structures from an organism, are essential genomic data, especially for phylogenomics and gene mining, for which accessing reliable protein sequences from publicly available emerging draft genomes is invaluable (Keeling and Burki, 2019). These can be more or less accurately obtained through the structural annotation of a genome, for which the collection of evidence data and the use of annotation pipelines are tricky at best (Yandell and Ence, 2012).
Following the decrease in sequencing costs due to the advent of Next Generation Sequencing and the concomitant explosion of sequenced organisms, new genomic data from emerging model organisms allow researchers to access unexplored taxonomic groups (Keeling and Burki, 2019). However, eukaryotic genomes, whose biodiversity is predominantly represented by protist lineages (Adl et al., 2019, Burki et al., 2020), present special features which complexify the structural annotation process: large genomes with a low gene density, long intergenic regions, as well as introns (Yandell and Ence, 2012). Although pipelines for eukaryotic genome annotation have been developed for more than a decade, it is still challenging to obtain an accurate annotation of the gene structures, a shortcoming that is often revealed in phylogenomic studies (Di Franco et al., 2019). MAKER2 (Holt and Yandell, 2011) has been, for more than a decade, one of the most popular annotation pipelines for eukaryotes.
Although MAKER2 (Holt and Yandell, 2011) enables individual laboratories to annotate non-model organisms (for which pre-existing gene models are not available), the use of this tool remains complex, as it implies the orchestration and fine-tuning of a multi-step process (Campbell et al., 2015). First, an evidence dataset must be compiled by collecting phylogenetically related proteins and species-specific transcripts, which often requires the assembly of RNA-Seq data for new organisms. Next, iterative runs of MAKER2 (Holt and Yandell, 2011) must also be coordinated to aim for accurate predictions, which includes intermediary specific training of different gene predictor models.
Here we present AMAW (Automated MAKER2 Annotation Wrapper) (Loïc Meunier et al., 2022), a wrapper pipeline facilitating the annotation of emerging unicellular eukaryotes (i.e., protist) genomes in both small and large-scale projects in a grid-computing environment. This tool addresses all the above-mentioned tasks according to MAKER2 authors’ recommendations (Campbell et al., 2015) and is, to our knowledge, the first implementation automating the use of MAKER2. We also demonstrate that the use of AMAW yields genome annotation significantly improved in comparison to the use of MAKER2 with the AUGUSTUS (Stanke et al., 2008) gene models that are available by default.
AMAW is implemented in Perl 5 version 22 (Perl, 1994) (RRID:SCR_018313) and is available either in a standalone version or through a Singularity container. Basic inputs required by AMAW pipeline are a FASTA-formatted nucleotide genome file and the organism name. Alternatively, evidence data, such as proteins or transcripts/ESTs provided by the user, or even gene models, can also be directly used for genome annotation.
The MAKER2 annotation suite was chosen to be automated for its performance and interesting features: beside supporting gene prediction with evidence data, MAKER2 has been demonstrated to improve the accuracy of its internal gene predictors, to maintain this accuracy even when the quality or size of evidence data decreases, as well as to limit the number of overpredictions (Holt and Yandell, 2011).
Taking MAKER2 as its internal engine, AMAW is able to gather and assemble RNA-Seq evidence, collect protein evidence, iteratively train the hidden Markov models (HMMs) of the predictors to yield the most accurate evidence-supported annotation possible without manual curation nor prior expertise of the organism (see AMAW subsection). Our tool, designed for non-model unicellular eukaryotic genomes, presents helpful applications in phylogenomics and comparative genomics. Indeed, some taxonomic lineages still lack high-quality genomic data (Burki et al., 2020), and filling these gaps would extend studies to these interesting groups.
The pipeline devised in AMAW (Figure 1) aims to reach three goals: (1) to achieve the most accurate annotation of a non-model genome without manual curation, (2) to automate the use of MAKER2 for supporting large-scale annotation projects, and (3) to simplify its installation and usage for users without a strong bioinformatics background.
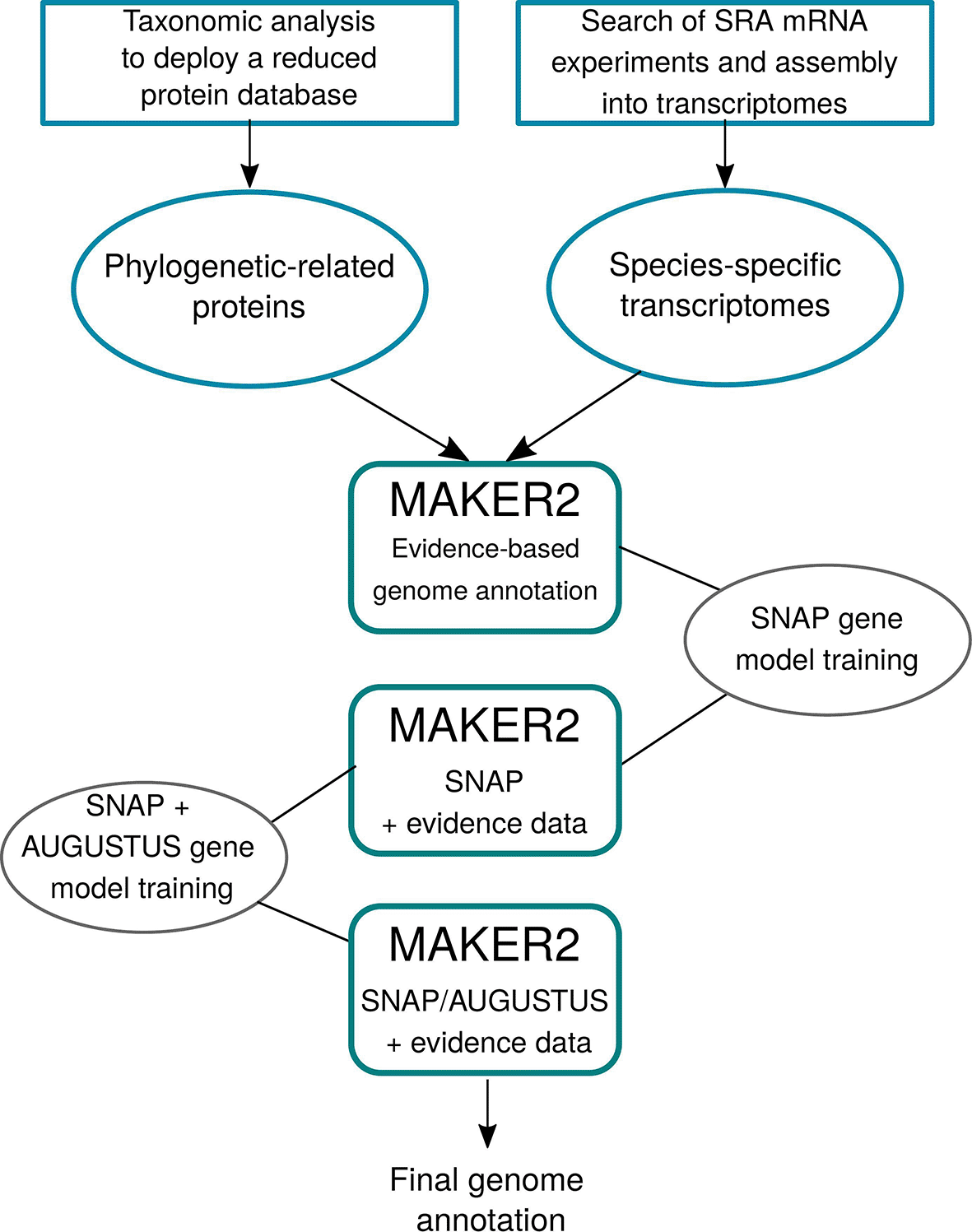
First transcripts and protein evidence are collected and deployed, if required. Then, three iterative runs of MAKER2 are performed to progressively train SNAP and AUGUSTUS gene predictors. The final genome annotation is generated after the third MAKER2 run.
First, a key factor for achieving accurate genome annotation is to collect as much evidence data (transcripts and/or proteins) as possible. This is needed both to optimize the training of specific gene models of ab initio gene predictors and to improve the confidence level in predictions supported by experimental data (Holt and Yandell, 2011).
Second, building evidence datasets is a time-consuming task, which also implies a certain level of bioinformatics skills. This consists of, in the best cases, finding and downloading directly available transcript or protein datasets for the genome species to annotate. However, this process often further requires assembling raw RNA-Seq reads into transcripts and gathering a reasonably sized protein dataset, usually including sequences of taxa phylogenetically close to the organism of interest. If building evidence datasets is feasible for a few genomes to annotate, doing so repeatedly for dozens or hundreds of genomes is hardly conceivable. This is why AMAW addresses this issue by automating the acquisition of both available RNA-sequence and protein data from reliable public databases (“NCBI Sequence Read Archive (SRA)” for RNA- sequence data and a combination of “Ensembl genomes” and NCBI databases for protein sequences).
Third, in addition of constructing a good input dataset for the annotation, AMAW automates the installation and the global use of the MAKER2 annotation pipeline based on good practices published by its authors (Campbell et al., 2015), and orchestrates the successive runs in a grid-computing environment. Even if MAKER2 is described as an easy to use pipeline, its handling and the optimal fine-tuning of its parameters demand that users take notice of its large documentation and, again, require a good bioinformatics understanding.
The complete workflow of AMAW can be summarized in three steps:
1. Transcript evidence data acquisition: RNA-Seq acquisition, assembly into transcripts, quantification of the abundance of the transcripts and filtering of redundant transcripts and minor isoforms;
2. Protein evidence deployment;
3. MAKER2 iterative runs and progressive training of its internal gene predictors.
It is possible for the user to provide their own in-house protein and/or transcript dataset(s). Moreover, they can short-circuit the pipeline by choosing an existing gene model for AUGUSTUS (Stanke et al., 2008) and/or SNAP (Korf, 2004). However, unless available models are well-suited for the organism at hand (matching species), it is advised to rely on AMAW full analysis.
Acquisition and building of transcript evidence data
The generation of a specific transcript dataset is carried out on the basis of the organism species name, provided by the user. This name is used to search for RNA-Seq experiments in NCBI SRA. Considering the divergence between nucleotide sequences at the genus level, only species-specific data is collected to perform direct nucleotide alignment (Campbell et al., 2015). The information of RNA-Seq experiment runs is collected with e-utilities and the corresponding FASTQ files are downloaded with fastq-dump v3.0.0. The acquisition of the RNA-Seq data prioritizes paired-end reads, when available, rather than single-end libraries, for more accurate transcript assembly. To limit the data volume to be stored in the case of well-represented organisms, two options are implemented: (1) a threshold on the maximal cumulative size of FASTQ files to download (by default: 25 GB) and (2) a threshold on the number of experiments (by default: none). Moreover, RNA-Seq experiments are sorted by ascending data volume before being selected in an attempt to maximize the diversity of RNA-Seq libraries.
FASTQ read files are assembled into transcripts with Trinity v2.12.0 (Grabherr et al., 2013) (standard parameters). The abundance of transcripts is first assessed with “align_and_estimate_abundance.pl”, a Trinity utility script that uses RSEM (Li and Dewey, 2011), then a custom script removes the redundant transcripts (which are common when several samples are pooled) and minor isoforms (by default, with abundance < 10% for a Trinity-defined gene). Finally, assembled transcripts are pooled and fetched to MAKER2.
Deployment of preloaded protein evidence data
To collect a set of curated protein sequences of eukaryotic microorganisms, Ensembl genomes (Kersey et al., 2018) were downloaded (Protists, Fungi and Plants - release 35.0, 08 May 2017) in combination with protist genomes available on the NCBI (March 2017) into a single database. However, to accelerate the computation time of MAKER2 annotations, this protein sequence database was subdivided following the major eukaryotic taxonomic clades. For this, we used the NCBI third taxonomic level (usually the phylum), which allows us to already considerably reduce the quantity of data to deploy for an annotation while ensuring enough sequence evidence for less studied lineages. Moreover, for further optimization of the computation time, these subsets were also dereplicated with CD-HIT version 4.6 (Li and Godzik, 2006): sequences sharing ≥ 99% identity were removed in favor of a single representative sequence. In practice, the taxonomy of the user-given organism species name is used to deploy the protein database corresponding to its taxon.
MAKER2 runs and intermediate trainings of the gene predictors
Following the good practices given by Campbell et al. (2015), the default AMAW workflow consists in three successive MAKER2 runs:
1. The first MAKER2 round predicts the genes only based on alignment of the provided transcript and protein data on the genome assembly to annotate. The predicted gene sequences will then be used for training a gene model for the SNAP gene predictor.
2. MAKER2 second round uses SNAP with the trained gene model and the evidence data will only be used for supporting the presence or absence of the predicted genes. Then, the SNAP gene model is trained again and a gene model is trained for AUGUSTUS.
3. MAKER2 third and last round performs gene predictions with both trained SNAP and AUGUSTUS gene predictors.
At the end of these three annotation rounds, two sets of gene predictions containing the gene predictors consensus are returned: a first one containing those supported by evidence data and a second one with the unsupported ones. However, the latter dataset needs to be cautiously used as the false positive rate is expected to be higher.
For optimal performance of the pipeline, it is possible (and recommended when applicable) for the user to provide her/his own experimental transcript data.
Beside the complete pipeline, AMAW also offers the possibility to shorten the analyses to only one round to:
- annotate several genomes of the same species (or re-run a previous analysis) for which the evidence data has already been constructed and the SNAP and AUGUSTUS gene models already trained.
- directly use an AUGUSTUS gene model (available in its library or provided by the user) without evidence data building. It is noteworthy that this mode does not use the SNAP gene predictor.
In this case, only the third round is launched according to the chosen mode.
The efficiency of MAKER2 being well known (Holt and Yandell, 2011), we illustrate the performance of AMAW by comparing its annotations with those of MAKER2 on a selection of 32 protist genomes in two very contrasted conditions (Cornet, Luc 2022a). In detail, the annotations generated with AMAW, where a gene model is specifically created for the genome from the available data, are compared with those produced with gene models directly available in AUGUSTUS (Stanke et al., 2008). The latter (control) condition corresponds to a basic usage of MAKER2.
To explore the impact of gene model choice, four AUGUSTUS models were used against AMAW generated ones: Homo sapiens, Arabidopsis thaliana, Aspergillus oryzae and the “closest” available model with respect to the organism to annotate. For this, a dataset of 32 genomes of protist organisms was designed and the quality of the different structural annotations was assessed using the completeness metrics provided by BUSCO v4 (Seppey et al., 2019) and the latest orthologous databases (Kriventseva et al., 2019). The genomes were downloaded from the NCBI and are available in the Supplementary Database (Cornet, Luc 2022a). For more details, see Supplementary Tables 1 (Cornet, Luc 2022e) and 2 (Cornet, Luc 2022f) for the complete taxonomy of these genomes, evidence data used to train the gene models and orthologous databases used with BUSCO.
The analysis of median values of BUSCO metrics shows that AMAW gene models significantly improve the quality of MAKER2 annotations (Figure 2A): with a median completeness of 90.6% (the closest gene model is the second most complete with a median of 68.7%), a median rate of fragmented annotations of 3.8% (second: closest gene model with 8.2%) and a median rate of missing annotations of 5.4% (second: closest gene model with 14.0%). Complete BUSCO results are provided as a table (see Supplementary Table 3 (Cornet, Luc 2022g)) and individual barplots for completeness, fragmented and missing genes (see Supplementary Figures 1 (Cornet, Luc 2022b), 2 (Cornet, Luc 2022c) and 3 (Cornet, Luc 2022d), respectively).
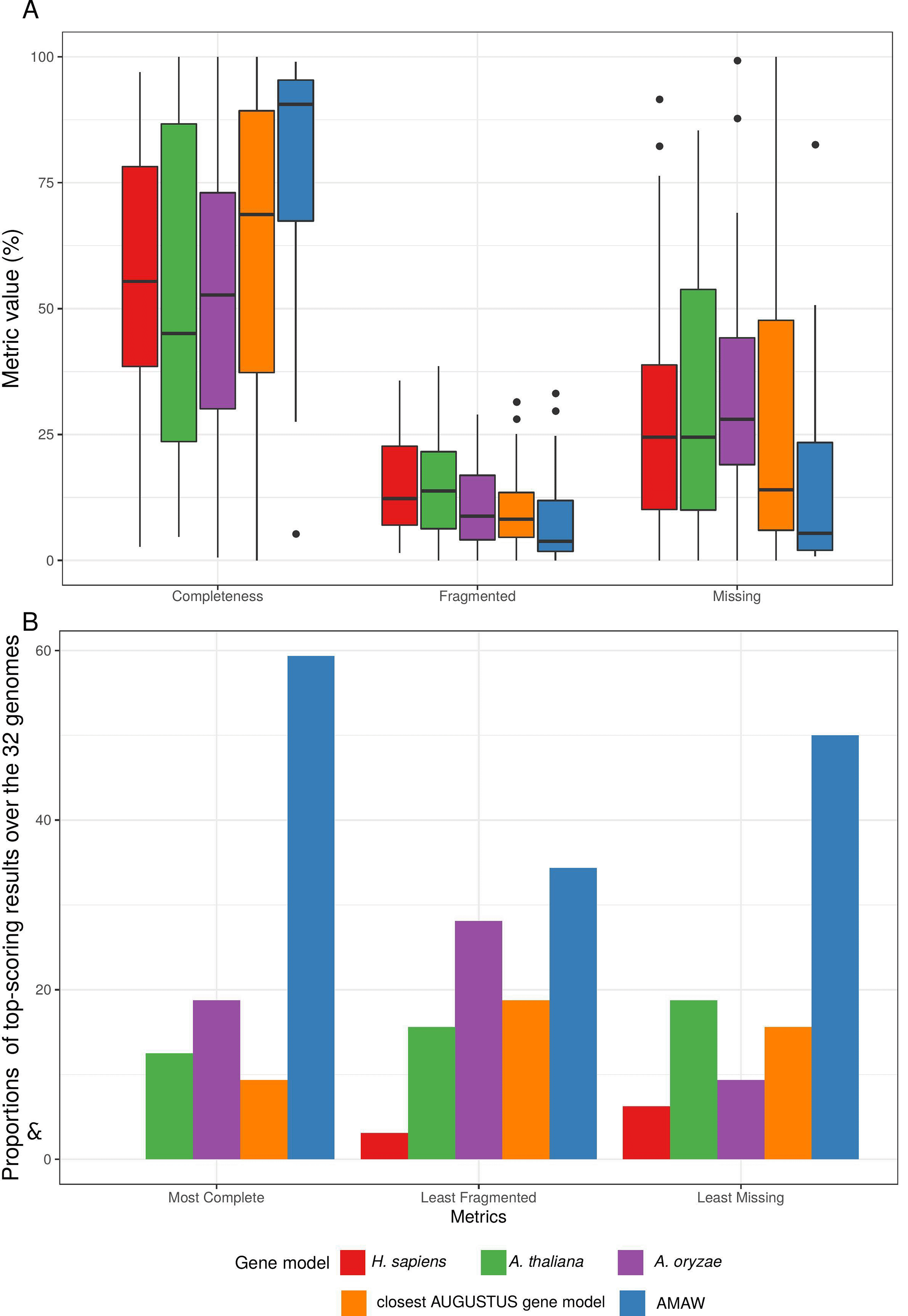
Among the five gene models used for each genome, AMAW performed best, giving the most complete annotation in 59.4% of cases, the least fragmented annotations in 34.4.8% of cases and the lowest proportion of missing proteins in 50.0% of cases (Figure 2B). AMAW annotations for which RNA-Seq data is available are of better quality (see Figure 3).
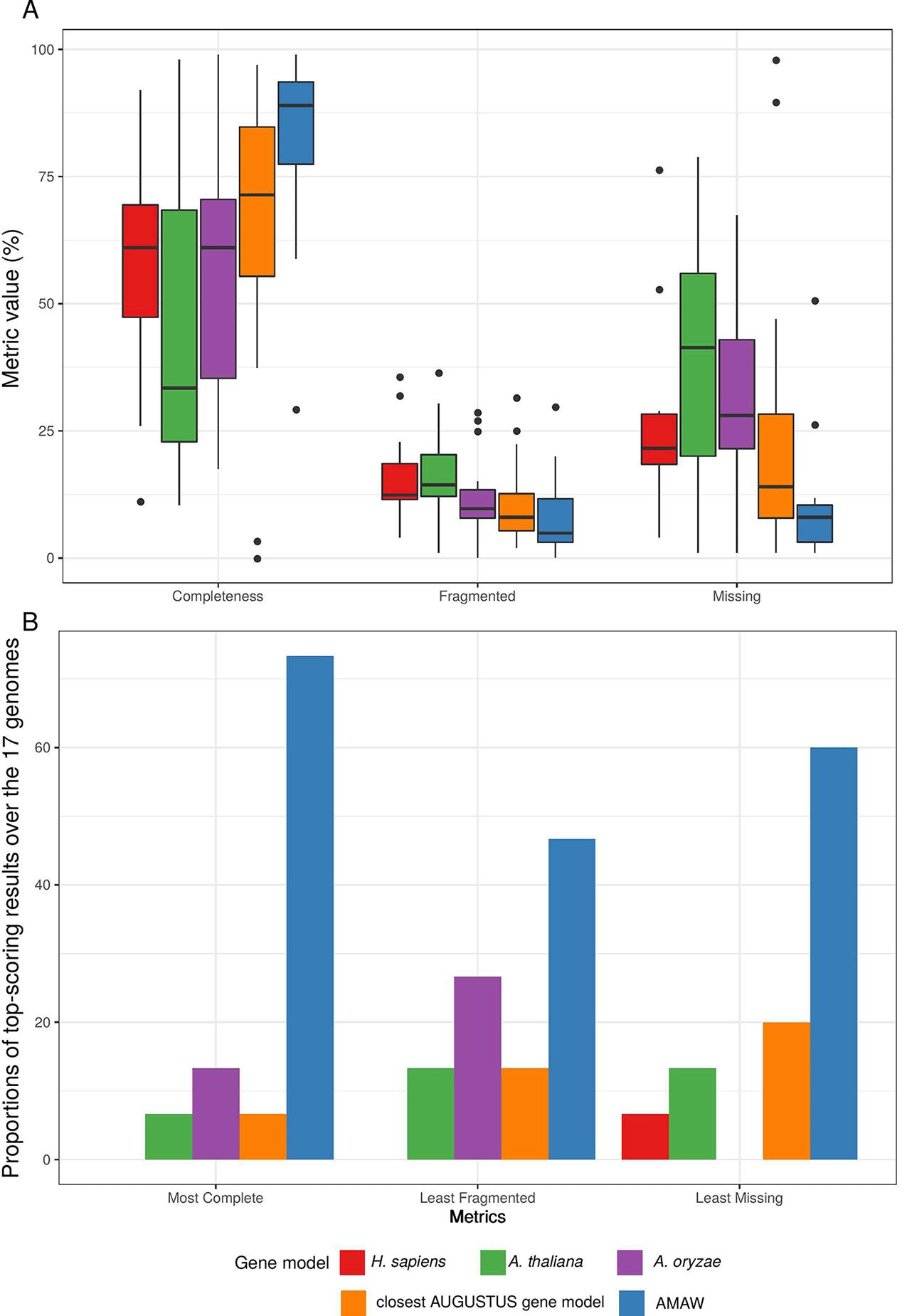
Among the five gene models assayed for each genome, AMAW performed best, giving the most complete annotation in 73.3% of cases (in comparison with 59.4% for the full genome dataset), the least fragmented annotations in 46.7% of cases (in comparison with 34.4%) and the lowest proportion of missing proteins in 60.0% of cases (in comparison with 50.0%).
We presented AMAW and its set of functionalities automazing the annotation of genomes, with a specific aim for non-model organisms. The application example shows how AMAW significantly improves the genome annotation quality in comparison of naive use of MAKER2 with pre-existing gene models, as well as the importance of providing specific evidence data. We aim with AMAW’s functionalities automating the acquisition and deployment of evidence data to contribute to the effort for achieving continually more complete and accurate annotations, especially for poorly represented eukaryotic lineages. Considering its streamlined installation and straightforward usage in grid-computing environments, we hope AMAW to be useful in future small and large genome annotation projects.
L. Meunier: Conceptualization, Data Curation, Formal Analysis, Investigation, Methodology, Software, Visualization, Writing - Original Draft Preparation.
D. Baurain: Conceptualization, Funding Acquisition, Methodology, Resources, Validation, Writing - Review & Editing.
L. Cornet: Conceptualization, Data Curation, Formal Analysis, Investigation, Software, Supervision, Writing - Review and Editing.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)