Keywords
ZINC database, SwissADME, MDR TB, fatty acid synthetase, ligand-based virtual screening.
This article is included in the Cheminformatics gateway.
This article is included in the Bioinformatics gateway.
ZINC database, SwissADME, MDR TB, fatty acid synthetase, ligand-based virtual screening.
According to World Health Organization (WHO, 2021), tuberculosis is caused by Mycobacterium tuberculosis and is spread through the air. Mycobacterium tuberculosis is thought to infect a quarter of the population and is more dangerous in malnourished people (WHO, 2021). A Mycobacterium strain resistant to at least Rifampicin and Isoniazid, the first-line medications used to treat all tuberculosis patients, causes multidrug drug-resistant TB (CDC, 2016). According to a study that was done by Seung et al., in 2015, it was found that multidrug-resistant TB has increased and has become a global health concern. MDR-TB affects over 470.000 people and kills roughly 180,000 yearly (WHO, 2021).
Most African countries still have gaps in diagnosis and treatment, resulting in a rise in MDR-TB patients. Only 68% of the individuals were found to have MDR-TB. MDR TB has emerged as a problem in Sub-Saharan Africa. The increase in MDR TB is due to a scarcity of appropriately trained health staff and resources for tuberculosis control (Ismail et al., 2018). In 2016, MDR TB was found to be 1.5% prevalent in Sub-Saharan Africa, with MDR cases increasing five times in Ethiopia for patients previously treated for TB compared to new cases (Musa et al., 2017). According to a recent study carried out in Mbagathi and Chandaria in 2015 and 2016, the prevalence of isoniazid resistance in Kenya was 0.8%, and 0.8% for multidrug resistance (Ogari et al., 2019). There has been less research in Kenya to illustrate the frequency of MDR TB. Therefore, there is insufficient data on the disease. There are also limited diagnostic tools, so detection is poor (Ombura et al., 2016).
The method of looking via computer databases for potentially active substances is known as virtual screening. There are two categories of virtual screening: ligand-based drug design and target-based drug design. Ligand-based drug design searches databases for compounds comparable to known ligands with similar biological actions (Neves et al., 2018). Virtual screening aids in the discovery of alternative compounds that perform the same biological function as MDR TB drugs but are more effective and less harmful (Banegas et al., 2018).
The study was carried out in August 2022. All of the medications were analyzed in silico. The canonical smiles of the drugs (pyrazinamide, ethambutol, cycloserine, bedaquiline, streptomycin, amikacin, kanamycin, linezolid, levofloxacin, and moxifloxacin) were obtained from PubChem (RRID:SCR_004284). The drugs were selected as they are the most common second-line agents that are used in the management of MDR TB. They are also easily found in hospitals.
The canonical smiles of the drugs were inserted into the Swiss Similarity online tool to identify other compounds that are similar to the drugs. Because it provides molecular shapes and properties, the Swiss similarity tool was used to determine molecular similarities. The combined technique of Swiss similarity that involves molecular fingerprints, pharmacophore recognition and shape-based similarity was used to screen the ZINC database of the drug-like compounds. Similarity scores were obtained based on the combination of Electro-shape 5D Manhattan distance and Tanimoto coefficient. 20 analogs that had the highest similarity scores to the reference compounds were selected and sketched on a PubChem sketcher, and the Molfile was downloaded and saved in an LBVS folder.
The drugs were downloaded in SDF format and saved in a drug-specific folder. Similar compound structures were relocated from the LBVS folder to the SBVS folder. The drugs and analogs’ Molfile were downloaded and transformed into their 3D structures using Avogadro version 1.1.0 software (Avogadro, n.d.) (RRID:SCR_015983). MMFF94S was then used to optimize the 3D structure to their most stable conformations.
The Chimera version 1.14c software (RRID:SCR_004097) was used to add hydrogen atoms and charge to the stable conformations of the compounds. The protein databank was then used to download the drugs’ respective target receptors (Fatty acid synthase, arabinosyltransferase, D-alanine L-racemase, F-ATP synthase subunit, 30S ribosomal RNA, 23S ribosomal RNA of 50S ribosomal subunit, DNA gyrase) which was saved in an SBVS folder. The Chimera software was used to remove the non-standard residues from the molecular target receptors and was saved as the final receptor. The surface binding of the selected compounds with the target receptors was done using the Auto dock vina (RRID:SCR_011958), found in the Chimera software. The surface binding of the drugs was also done to act as a positive control. Ligand interactions were then examined using discovery studio version 19.1.0.18287 software (RRID:SCR_015651).
The SWISS ADME online tool, which predicts drug and medicinal chemistry similarity and pharmacokinetic features, was used to forecast the pharmacokinetic profile of the compounds. The selected compounds’ pharmacokinetic parameters were entered into the SWISS ADME, and the compounds’ absorption, distribution, metabolism, and excretion properties were obtained.
The ProTox RRID:SCR_018506 server was utilized to predict the toxic levels of the medications and comparable substances. The canonical smiles of the drugs and the analogs were obtained from pubchem and pasted on ProTox server. Oral toxicity, immunotoxicity, carcinogenicity, cytotoxicity, and mutagenicity were all determined using the ProTox server. In determining oral toxicity, class 1 means it’s fatal if swallowed (LD50 less than 50 mg/kg), class II fatal if swallowed with an LD50 of between 5 mg/kg to 50 mg/kg, class III is toxic when swallowed with an LD50 of between 50 mg/kg to 300 mg/kg, class IV is harmful wen swallowed with and LD50 of between 300 mg/kg to 2000 mg/kg, class V may be harmful when swallowed with an LD50 of between 2000 mg/kg to 5000 mg/kg and class VI is non-toxic with an LD50 of greater than 5000 mg/kg.
Cycloserine had a docking score of -4.6, and the analogs had higher binding energies than cycloserine (Muturia, 2023). The analogs of cycloserine that were chosen had a predicted toxicity class of class V as shown in Table 1, which indicated that they were less hazardous. All substances lacked CYP1A2 inhibition, CYP3A4 inhibition, CYP2D6 inhibition, CYP2C19 inhibition, BBB penetration, and a p-glycoprotein substrate. Except for ZINC3906759, all substances were anticipated to have a high GI absorption. ZINC9592220, ZINC95922219, and ZINC39067759 were predicted to be active for mutagenicity and carcinogenicity. The interaction of cycloserine analog ZINC39067759 with D-alanine is due to van der Waals, unfavorable charges and attractive charges that indicate possible ligand and protein interactions as shown in Figure 1.
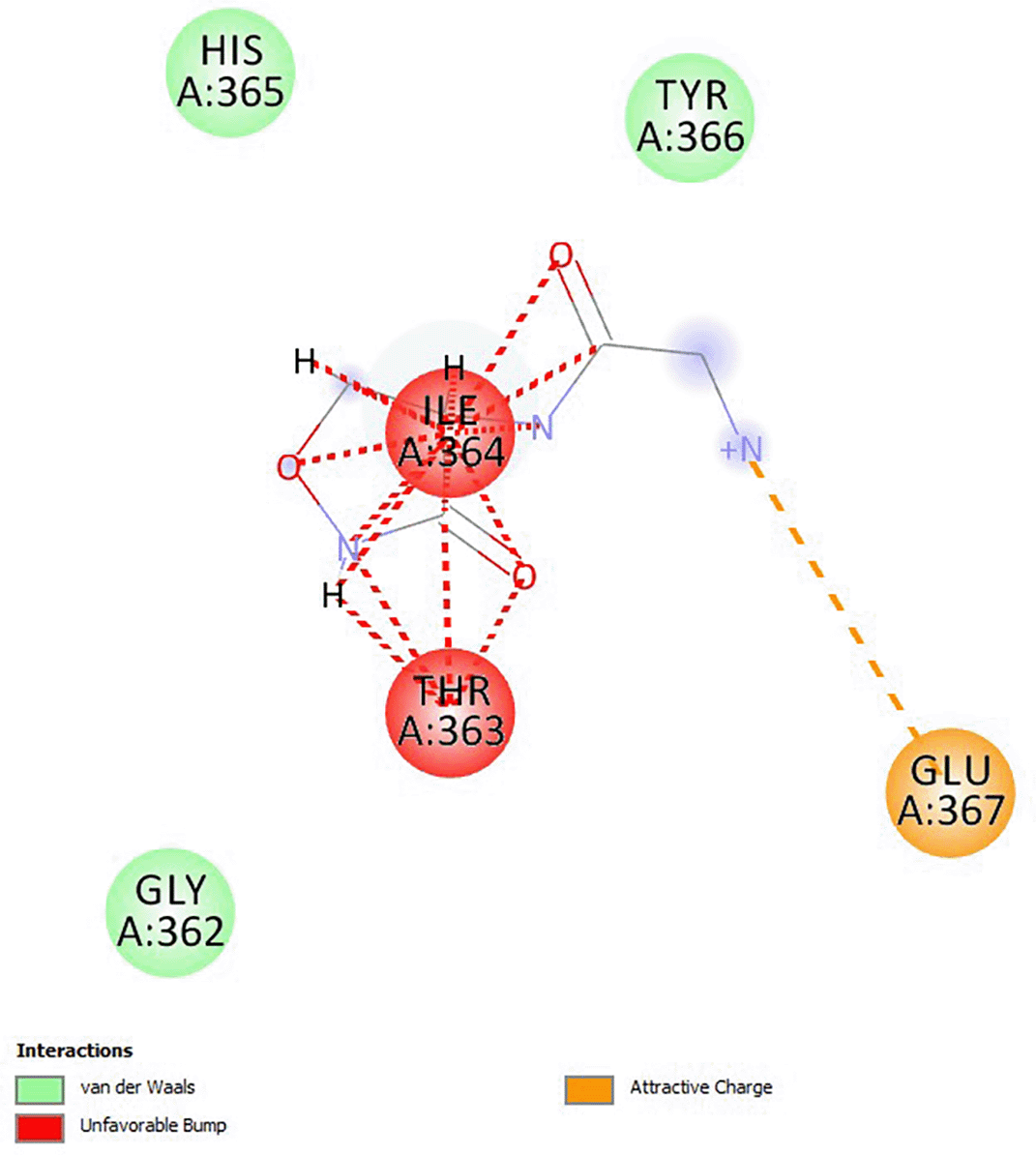
Source: this study. The diagram was created by discovery studio software.
The docking score for amikacin was 5.5. In comparison to amikacin, two of the chosen analogs had binding energies that were the same, one had greater binding energy, and seven had lower binding energies. Amikacin did not follow the Lipinski rule of five. Amikacin belonged to class V, while the chosen analogs were in classes V and VI as indicated in Table 1. All of the substances demonstrated minimal GI absorption and no CYP1A, CYP2C9, CYP2C19, or CYP3A4 inhibition. Except for ZINC44401823, all substances had a p-glycoprotein substrate. The interaction of amikacin with the 30s ribosomal subunit is due to van der Waals forces, carbon-hydrogen bonds, unfavorable positive-positive charges as well as conventional hydrogen bonds as shown in Figure 2.
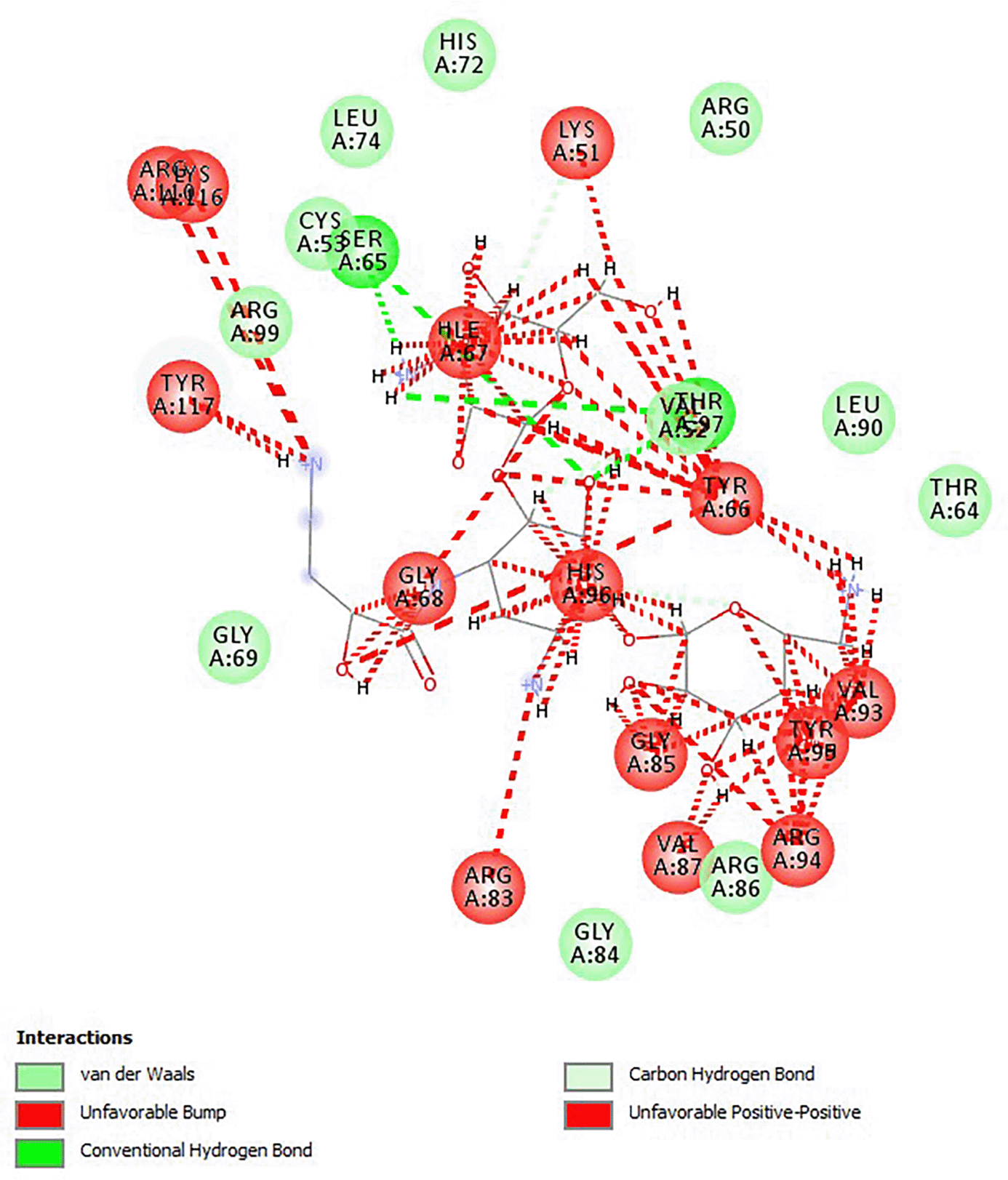
Source: this study.
The binding energies of the analogs were higher than that of pyrazinamide, which had a binding energy of -4.9. Except for ZINC95709439 and ZINC00351669, which are in toxicity class V, all compounds had a predicted toxicity level of IV as shown in Table 1. None of the substances inhibited CYP1A2, CYP2C9, CYP3A4, or CYP2C19, permeated the blood-brain barrier, or served as a p-glycoprotein substrate. Additionally, their GI absorption was high. Both ZINC95709439 and ZINC06524479 were active for hepatotoxicity and mutagenicity, respectively. Mutagenicity and immunotoxicity were both active for ZINC66351878 and ZINC61508077, respectively. The interaction of pyrazinamide and fatty acid synthetase is due to pi-alkyl, conventional hydrogen bond, and van der Waals forces as shown in Figure 3.
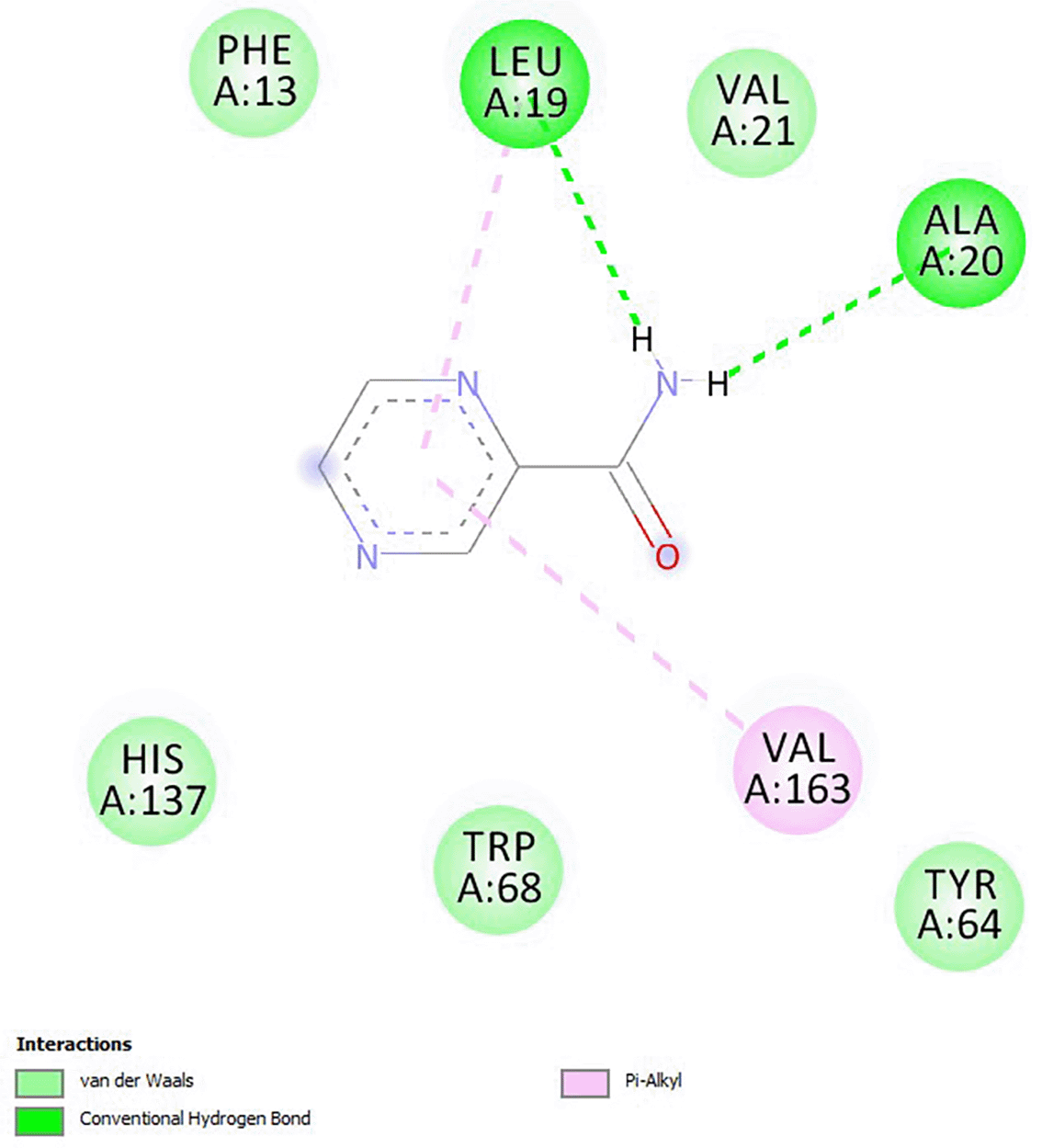
Source: this study.
Ethambutol had a binding energy of -4.7, and all of its analogs had greater binding energy. All substances lacked BBB penetration, p-glycoprotein substrate, CYP1A2 inhibition, CYP2C19 inhibition, CYP2C9 inhibition, and CYP3A4 inhibition. Ethambutol belongs to toxicity class IV. As indicated in Table 1, the predicted toxicity class for four of the analogs was V, whereas the predicted toxicity class for the other six analogs was VI. Except for ZINC23477159, ZINC19364558, ZINC2247967, and ZINC22947968, all compounds showed a high GI absorption. The carcinogenicity of ZINC19365151, ZINC19364877, ZINC19361997, and ZINC100648389 and ZINC19365150 was active. The interaction of ethambutol and arabinosyl transferase is due to van der Waal’s forces, attractive charges, conventional hydrogen board, carbon-hydrogen board, alkyl, pi-alkyl, and unfavorable positive-positive charges as shown in Figure 4.
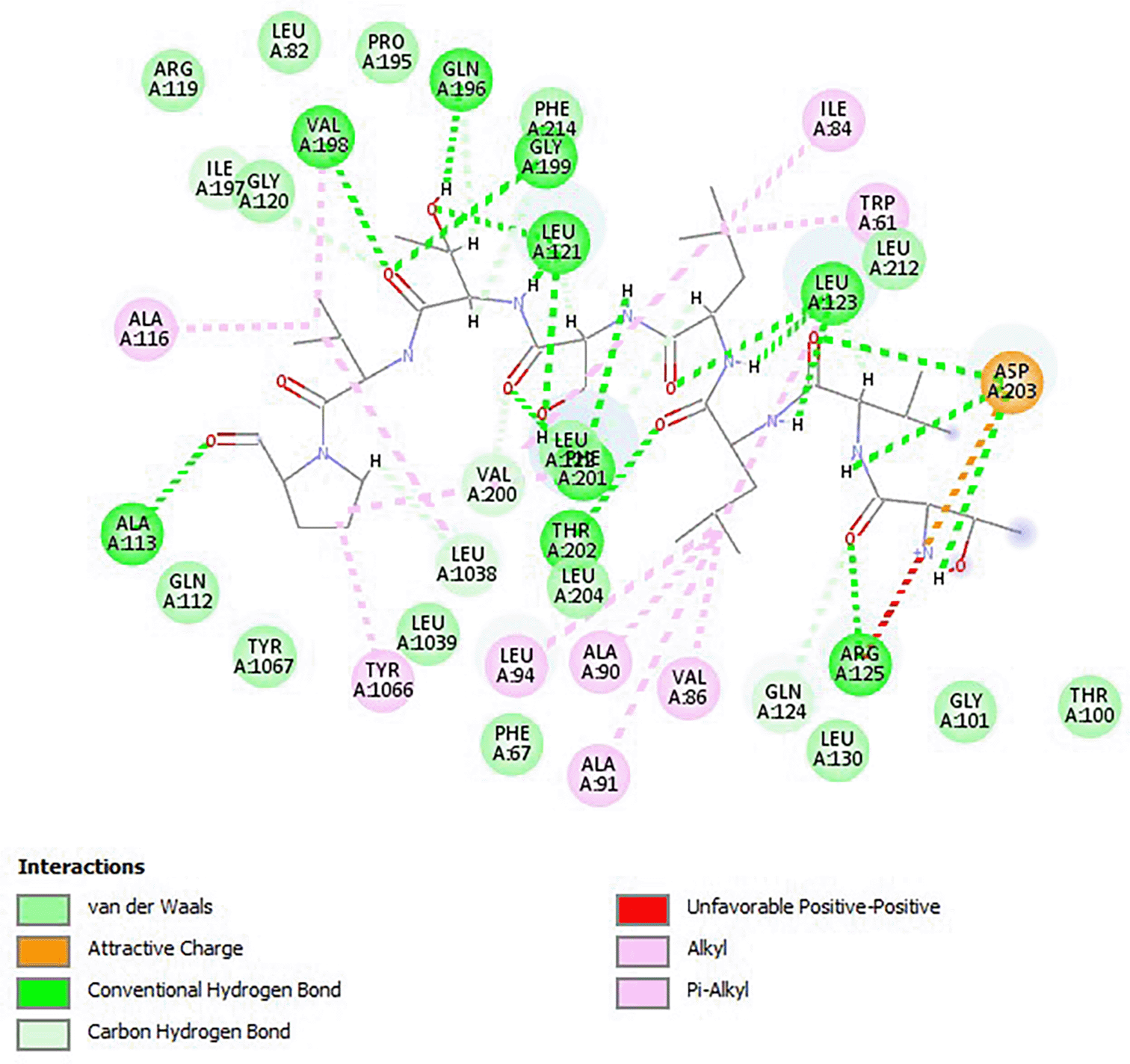
Source: this study.
The SwissSimilarity tool was used to find substitute compounds that were similar to the medications used to treat MDR TB. From each medication, 20 analogs were chosen. The 20 analogs that were selected were based on the highest similarity scores to the reference compounds. The comparison of the docking energies was used to identify compounds with the highest docking energies as these are the compounds with the best affinity to the receptors. Out of the 200 analogs chosen, 98 had higher binding energies, 88 had lower docking scores, and 14 had the same docking scores as the primary medication. 98 compounds would have been chosen because they had the best docking scores if we had only looked at docking scores. 20 of the 98 analogs were for pyrazinamide, 20 were for cycloserine, and 20 were for ethambutol. Meaning all of their analogs had the best docking scores and hence are potential compounds that could undergo further studies compared to the other compounds.
According to the Lipinski rule of drug-likeness, the analogs of bedaquiline, cycloserine, moxifloxacin, ethambutol, pyrazinamide, levofloxacin, and linezolid all had molecular weights less than 500, hydrogen acceptors less than 10, log P less than 5, and hydrogen bonds less than 5, making them all orally active (Daina et al., 2017). Since they did not adhere to the Lipinski rule, streptomycin, amikacin, kanamycin, their analogs, and bedaquiline are not orally active (Daina et al., 2017). Class I toxicity have extremely lethal effects when ingested and has an LD50 of less than 5 mg/kg. Class III toxicity has an LD50 range of 50 mg/kg to 300 mg/kg and may be hazardous when ingested, while class II toxicity has an LD50 that ranges from 5 mg/kg to 50 mg/kg and are lethal when ingested. Class IV has a potential for harm when ingested and has an LD50 range of 300 to 2000 mg/kg. Class V is less dangerous and can be consumed by humans without having negative effects because it has an LD50 value of more than 2000. Class VI is non-toxic and has an LD50 of more than 5000, therefore humans can consume it without experiencing any negative consequences (Gadaleta et al., 2019).
Cycloserine works by preventing the peptidoglycans from forming, which weakens the cell wall and causes the bacteria to die. When used orally, it has a high GI absorption of between 70% and 90% (Wishart et al., 2018). It is necessary to lower the doses for people who have renal toxicity (Goodman & Gilman, 2018). Cycloserine has several negative side effects, including anxiety, mental distress, nervousness, nightmares, muscular twitching, and vertigo (Wishart et al., 2018). Consuming cycloserine with fattening foods reduces its absorption (Wishart et al., 2018).
Amikacin, generated from kanamycin A, attaches to the 30S ribosomal subunit and prevents mRNA from binding to it and tRNA from accepting it, preventing bacterial growth (NCBI, 2022a). When injected intramuscularly, it has a high absorption rate, but topical and oral absorption is poor (NCBI, 2022a). It is eliminated by the kidney and may cause neuromuscular blockade, nephrotoxicity, and ototoxicity as side effects (Wishart et al., 2018).
According to National Center for Biotechnology Information (2022b), pyrazinamide inhibits fatty acid synthetase 1, which prevents the bacteria from producing the fatty acids necessary for growth and replication, leading to the death of the bacteria. It enters the GI system quickly and is 10% bound to plasma proteins. At least 70% of the oral dose is eliminated in the urine, primarily by glomerular filtration, and it is subject to hepatic metabolism (NCBI, 2022b). Adverse effects may be stomach upset, fatigue, easy bruising, skin rash, yellowing of the skin or eyes, liver injuries, arthralgia, malaise, and urticaria. When taken with abacavir, pyrazinamide may reduce the drug’s elimination, which could raise the drug’s serum levels and cause toxicity (Wishart et al., 2018). Acetaminophen and Aceclofenac may reduce the elimination rate of pyrazinamide, increasing its concentration and causing toxicity (Wishart et al., 2018).
Ethambutol, a bacteriostatic drug, was created to treat Mycobacterium tuberculosis strains that were resistant to the antibiotic isoniazid. It prevents the synthesis of cell wall constituents like arabinogalactan and lipoarabinomannan by inhibiting arabinosyltransferases. Inhibiting the production of lipoarabinomannan prevents mycobacterial cells from interacting with the host cells. It has an oral bioavailability of between 75% and 80% (Wishart et al., 2018).
Ethambutol has a plasma binding of 20% to 30% and is metabolized by aldehyde dehydrogenase. Ethambutol is eliminated in urine in two forms: unmodified in 50% of cases and inactive metabolites in 8% to 15% of cases. Optic neuropathy, joint discomfort, pruritus, abdominal pain, malaise, and dizziness are some undesirable symptoms that may be noticed. Antacids shouldn’t be taken with the medication because they will stop Ethambutol from being absorbed (Wishart et al., 2018).
The discovery of new pharmaceuticals that may be utilized to treat multidrug-resistant tuberculosis has been made possible by this research’s contribution to the identification of compounds with higher docking scores than conventional treatments. Patients with other comorbidities will benefit from compounds without cytochrome inhibition since they have a lower risk of generating drug-drug or drug-herb interactions.
The potential compounds that were found were; ZINC01568476, ZINC01568479, ZINC96330915, ZINC96330916, ZINC01568477, ZINC01568478, ZINC05131962, ZINC44401823, ZINC00351669, ZINC19364232, ZINC19364558, ZINC23477159, ZINC22947967 and ZINC22947968 as shown in Table 1. These are compounds that had better binding energies compared to the standard drug, lacked CYP inhibition, lacked substrate for P-glycoprotein, and were in toxicity class V or VI.
Additional in vivo and in vitro research should be carried out on the following potential compounds; ZINC01568476, ZINC01568479, ZINC96330915, ZINC96330916, ZINC01568477, ZINC01568478, ZINC05131962, ZINC44401823, ZINC00351669, ZINC19364232, ZINC19364558, ZINC23477159, ZINC22947967, and ZINC22947968.
The analogs can also be tested for other indications where the drugs, ethambutol, streptomycin, pyrazinamide, amikacin, and cycloserine were used apart from multidrug-resistant tuberculosis.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)