Keywords
amplicon sequencing, data visualization, summary report, R package
This article is included in the RPackage gateway.
This article is included in the Bioinformatics gateway.
amplicon sequencing, data visualization, summary report, R package
Amplicon sequencing (AmpSeq) is increasingly used in biological and medical studies for its high depth of coverage, allowing identification of rare variants, and also the ability to multiplex hundreds of samples in a single run, thereby reducing the sequencing costs. AmpSeq is highly scalable and cheap, permitting low-cost data generation for large sample sizes. It can also be multiplexed to further increase resolution. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of coronavirus disease 2019 (COVID-19), continues to pose a serious threat to the global population health. AmpSeq has been widely used to target the SARS-CoV-2 viral genome regions to monitor the presence of SARS-CoV-2 and detect the emerging variants.1,2 AmpSeq methods are also widely used in infectious disease characterization and surveillance studies (e.g., malaria infection) to monitor the emergence and spread of drug resistance,3–7 to examine population structure,8,9 relapse,10 to estimate clearance rates11 and to track within-host dynamics in longitudinal studies.12
There are several bioinformatics pipelines for processing AmpSeq data, such as DADA2,13 SeekDeep,14 and HaplotypR.15 SeekDeep and HaplotypR were specifically developed and tailored to study Plasmodium spp. However, these pipelines have some limitations, such as a lack of downstream analysis, difficulty in distinguishing between true sequence and PCR sequencing errors and artifacts, and often requiring extensive bioinformatics expertise. We introduce AmpSeqR, a freely available R package (https://github.com/bahlolab/AmpSeqR) that performs AmpSeq data analysis. The pipeline integrates several R packages as well as newly developed functions to filter out sequencing noise and improve the accuracy of the detected sequence data. The pipeline offers various analysis steps including data pre-processing, amplicon sequence variants (ASVs) estimation, data post-processing, data visualization, and automatically generates a comprehensive report in Rmarkdown that contains all essential results. This pipeline is designed to simplify bioinformatics processing leading to a comprehensive pipeline that starts from raw FASTQ files and finishes by generating a final reproducible report, from a reproducible pipeline. We apply AmpSeqR to various AmpSeq datasets, including the SARS-CoV-2 dataset and malaria parasite Plasmodium falciparum datasets.
The AmpSeqR package was built in R and is hosted on GitHub. It can be installed from https://github.com/bahlolab/AmpSeqR. The input files for AmpSeqR are the standard paired-end FASTQ format provided by the common Illumina sequencing platforms (e.g., MiSeq), as well as sample barcodes and target amplicon details.
AmpSeqR was developed and tested on R (version 4.1.3) with several other dependencies and the base functions in R and is compatible with Mac OS X, Windows, and major Linux operating systems. AmpSeqR is maintained at GitHub (https://github.com/bahlolab/AmpSeqR). The archived source code can be found in https://doi.org/10.5281/zenodo.7580184.16 AmpSeqR uses Biostrings17 (v2.62.0, RRID:SCR_016949), ShortRead18 (v1.52.0, RRID:SCR_006813), DADA213 (v1.20.0), DECIPHER19 (v2.20.0, RRID:SCR_006552), rlang (v1.0.2), Rmarkdown (v2.13), gtools (v3.9.2), withr (v2.5.0), utils (v4.1.1), cowplot (v1.1.1), furrr (v0.2.3), future (v1.24.0), GenomicRanges20 (v1.46.1), VariantAnnotation21 (v1.38.0), S4Vectors (v0.32.3), IRanges20 (v2.28.0), plyr (v1.8.7), DT (v0.22), plotly (v4.10.0, RRID:SCR_013991), viridisLite (v0.4.0), flextable (v0.7.0), scales (v1.2.0, RRID:SCR_019295), magrittr (v2.0.3), heatmaply (v1.3.0), digest (v0.6.29), tidyselect (v1.1.2), data.table (v1.14.2), ComplexHeatmap22 (v2.11.1, RRID:SCR_017270), htmltools (v0.5.2), ape (v5.6-2, RRID:SCR_017343), randomcoloR (v1.1.0.1), ggtree23 (v3.0.4, RRID:SCR_018560), ggstance (v0.3.5), tibble (v3.1.8), stats (v4.1.3) and several tidyverse packages (v1.3.1, RRID:SCR_019186). The workflow is illustrated in Figure 1.
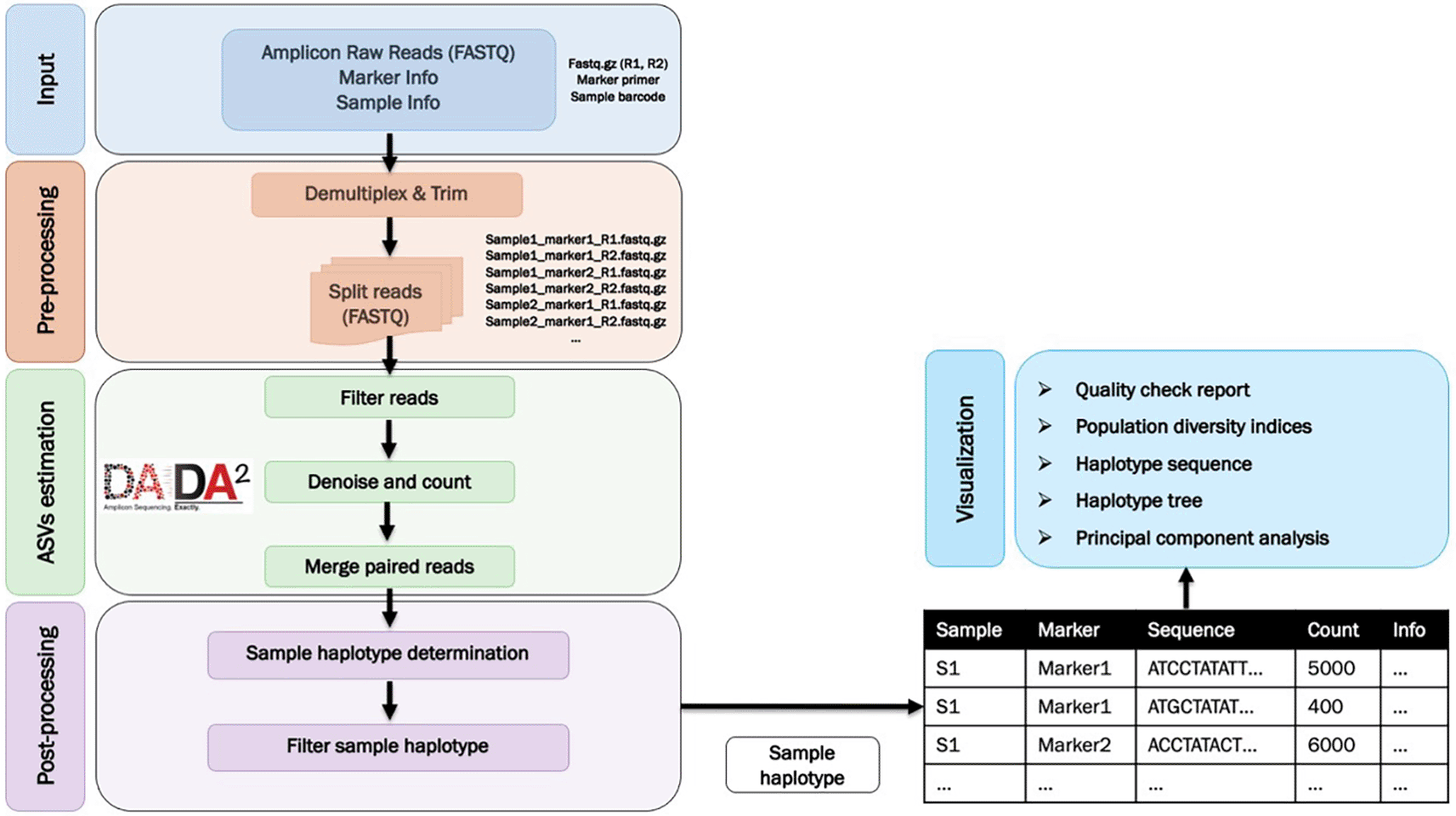
Data pre-processing
The data pre-processing step demultiplexes the raw paired-end FASTQ reads and trims the sample barcodes and target amplicon primer sequences as well as assigning the sequence to each sample and marker (amplicon target) combination. As each individual has a unique oligonucleotide barcode and each marker has a unique primer sequence distinguishing it from other markers, sequence reads can be de-multiplexed to separate individual samples and different markers, followed by trimming the primer sequence. The data pre-processing is handled using the Biostrings, ShortRead, rlang, magrittr, future, and tidyverse R packages.
Amplicon sequence variants (ASVs) estimation
The amplicon sequence variants (ASVs) estimation process identifies amplicon haplotype sequences. We leveraged several functions in the DADA2 R package, such as filterAndTrim, derepFastq, learnErrors, dada, and mergePairs to obtain the haplotype sequence. DADA2 can handle sequences with indels and accurately determines sequence variants leading us to choose DADA2 as the preprocessing tool to call ASVs in our AmpSeqR pipeline. For merging of paired-end reads we provided an option that can process the paired-end reads without any overlap. We also added some functions based on ShortRead, future, and tidyverse R packages for automatic downsampling of each sample/amplicon combination to a certain number of reads (e.g., 10,000) to reduce the computation time required. This step reduces excessive coverage and computational cost. The GATK (RRID:SCR_001876) documentation states that having additional data is not informative when read coverage exceeds a certain depth. A coverage of 10,000 reads was found to be sufficient to detect minor clones with a frequency as low as 0.1%.15
Data post-processing
This step primarily optimises amplicon haplotype calling through chimeric read detection and removal and variant filtration. The removal of sequencing background noise such as ultra-rare variants and chimeric reads generated during PCR amplification are important steps for the generation of high fidelity amplicon sequencing data. Chimeric reads are sequences formed from two or more biologically distinct sequences joined together during PCR amplification. Chimeric reads are much more frequent in amplicon sequencing applications in comparison to whole-exome sequencing (WES) or whole genome sequencing (WGS) because the same as well as very similar sequences appear in increased abundance.24 In addition, there are several types of indel errors in short-read Illumina sequencing data, such as homopolymers and terminal indels, which are the main source of low-quality indel calls.25 Several parameters are used to filter haplotypes, such as: (i) haplotype frequency, (ii) sequence similarity (haplotype sequences were mapped against reference sequence for each amplicon marker to calculate the sequence similarity), as well as (iii) variant heterozygosity, and (iv) minor allele frequency (MAF) in a given dataset. The detailed parameter options and their default values are listed in Table 1. After removing the background noise, the final haplotypes for each sample and marker combination are identified. This step is handled using the Biostrings, DECIPHER, rlang, withr, GenomicRanges20 (RRID:SCR_000025), VariantAnnotation21 (RRID:SCR_000074), S4Vectors, IRanges20 (RRID:SCR_006420), Parallel, gtools, utils, magrittr, tibble, and tidyverse R packages.
Data visualization
The data visualization step generates a comprehensive Rmarkdown report that contains various summary data visualizations and data summaries, such as quality checks, performed at both the individual/sample and amplicon level, amplicon haplotype sequence counts, haplotype diversity metrics of amplicon markers in the dataset, haplotype sequence visualization, haplotype tree generation, and principal component analysis (PCA). Quality checks include checking sample and amplicon marker information, an overview of read counts to track reads at various points in the pipeline, missing values in the dataset, and passed samples and markers. Haplotype diversity metrics such as expected heterozygosity (He), number of single nucleotide polymorphisms (SNPs), and number of unique haplotypes detected per sample per marker, are generated to help explore the amplicon marker diversity in the dataset. Detected haplotype sequences can be aligned to the reference genome and visualized to explore SNP positions and nucleotide changes. The haplotype tree is created by integrating all major haplotype sequences (>50% relative abundance) in each sample and performing multiple sequence alignment to identify the similarity between samples. PCA is also performed to visualize the sample structure. The process is handled using the Biostrings, Rmarkdown, plyr, DT, plotly, viridisLite, stats, flextable, ComplexHeatmap,22 scales, heatmaply, digest, tidyselect, data.table, DECIPHER, htmltools, pheatmap (RRID:SCR_016418), ape, randomcoloR, ggtree,23 ggstance, and several tidyverse R packages.
The major functions of the AmpSeqR package are summarised in Table 2.
In this section, we demonstrate the application of AmpSeqR for the data referenced in Datasets.
Three datasets are used for the Use cases section. Dataset 1 is provided with the package and datasets 2 and 3 need to be downloaded via the published papers in which they appeared.
Dataset 1: Synthetic Plasmodium falciparum AmpSeq data
The synthetic P. falciparum paired-end amplicon sequencing data (FASTQ) was generated using the ART26 and ShortRead18 packages. The dataset consists of five P. falciparum samples with defined mixtures (1:1, 1:10, 1:100, 1:500, 1:1000) of two P. falciparum reference genomes Pf3D7 and PfDd2 (downloaded from PlasmoDB). We selected two amplicon markers, the P. falciparum surface marker genes circumsporozoite protein (CSP) and thrombospondin-related anonymous protein (TRAP). The FASTQ sequence data, as well as additional amplicon primer and sample barcode details are included in the AmpSeqR package.
Dataset 2: SARS-CoV-2 AmpSeq data
The dataset for multiplexed SARS-CoV-2 was obtained through the Aynaud et al.1 study. We downloaded the PoC cohort, which includes 19 nasopharyngeal swab samples, of which 17 were positive for SARS-CoV-2. This study targeted six amplicon markers: one human gene Peptidylprolyl Isomerase B (PPIB) as a quality control, and five SARS-CoV-2 regions targeting the Nucleocapsid (N), Envelope (E), RNA-dependent RNA polymerase (RdRP), and two regions of the Spike (S) gene that correspond to the receptor-binding domain (RBD) and the polybasic cleavage site (PBS). Amplicon primer details are given in Aynaud et al.1 Sequencing was performed using the Illumina MiSeq sequencing platform. This dataset and metadata are available in Gene Expression Omnibus (GEO) at NCBI (GEO accession number GSE160031). The complete genome SARS-CoV-2 isolate Wuhan-Hu-1 (NC_045512) was used as the reference genome for sequence analysis and was downloaded from NCBI (NC_045512.2).
Dataset 3: Plasmodium falciparum AmpSeq data
The P. falciparum dataset can be downloaded from the Lerch et al.15 study. The dataset consists of 16 defined mixtures of P. falciparum genomic DNA samples, where the mixtures of two P. falciparum lab strains HB3 and 3D7 were combined in various proportions (1:1, 1:10, 1:50, 1:100, 1:500, 1:1000, 1:1500, and 1:3000). Lerch et al. targeted two P. falciparum surface marker genes: (i) the conserved Plasmodium membrane protein (CPMP) and, (ii) the circumsporozite protein (CSP). This dataset is available on the NCBI-SRA website: BioProject accession PRJNA381546. Amplicon primer and sample barcodes details are given in the Lerch et al. study and can be downloaded from https://github.com/lerch-a/HaplotypR.git. This provides a test dataset to evaluate the detectability of minority clones of AmpseqR.
The AmpSeqR package is hosted in GitHub and can be installed with the following command line.
devtools::install_github("bahlolab/AmpSeqR")Load the AmpSeqR package into the workspace as well as other packages required for the following analysis.
library(AmpSeqR) library(tidyverse)
Here we present the AmpSeqR workflow with a minimal example dataset (example_data). The input files are the standard paired-end FASTQ files provided by the Illumina sequencing platforms, as well as sample barcodes and target amplicon details. The sample barcodes file should include sample_id, barcode_fwd, barcode_rev, sample (sample name, can be the same as sample_id), info (e.g., sample type). The target amplicon detail file should include marker_id, primer_fwd, primer_rev, seq (reference sequence), chrom (chromosome), start (reference sequence start position), end (reference sequence end position). This file should be set up in a text editor or excel and saved as text CSV format (*.csv) file.
example_data <- get_ampseqr_example_data()
example_data
## $marker_info
## # A tibble: 2 × 7
## marker_id primer_fwd primer_rev seq chrom start end
## <chr> <chr> <chr> <chr> <chr> <int> <dbl>
## 1 CSP GTGGAGTATGTCCGTAACT… CTAGCA… CCCA… Pf3D… 222253 222372
## 2 TRAP CTTCTACGTCTTACAAAGG… CGTGAT… TGGG… Pf3D… 1465085 1465204
##
## $sample_manifest
## # A tibble: 5 × 5
## sample_id barcode_fwd barcode_rev sample info
## <chr> <chr> <chr> <chr> <chr>
## 1 S01 ATCGCTAT TCTAATGT 3D7_Dd2_1_1 1_1
## 2 S02 GTGACGAA TTACAGCA 3D7_Dd2_1_10 1_10
## 3 S03 CTATTGCA GTGCTAAT 3D7_Dd2_1_100 1_100
## 4 S04 CGATCGAT GCACTTGT 3D7_Dd2_1_500 1_500
## 5 S05 AGAGGGAC GCAAGCGT 3D7_Dd2_1_1000 1_1000
##
## $reads_2
## [1] "~/R/x86_64-pc-linux-gnu-library/4.1/AmpSeqR/extdata/readsR.fastq.gz"
##
## $reads_1
## [1] "~/R/x86_64-pc-linux-gnu-library/4.1/AmpSeqR/extdata/readsF.fastq.gz"
##
## $data_dir
## [1] "~/R/x86_64-pc-linux-gnu-library/4.1/AmpSeqR/extdata"This step demultiplexes the raw paired-end FASTQ reads and trims the sample barcodes and target amplicon primer sequences and assigns reads to each sample and each amplicon marker combination.
Here, we first use the synthetic Plasmodium falciparum AmpSeq data.
# Input data reads_1 <- example_data$reads_1 reads_2 <- example_data$reads_2 sample_manifest <- example_data$sample_manifest marker_info <- example_data$marker_info
The analysis directory is then created.
# Create the analysis directory dir.create("./runs") run_dir <- "runs"
Next, we demultiplex the reads using the demultiplex_reads function as follows.
# Demultiplex reads demultiplexed <- demultiplex_reads(sample_manifest = sample_manifest, marker_info = marker_info, reads_1 = reads_1, reads_2 = reads_2, output_dir = run_dir, output_sub_dir = file.path (run_dir, 'demultiplex')) ## note: 100000 of 100000 reads demultiplexed successfully. demultiplexed ## # A tibble: 10 × 7 ## sample_id marker_id reads_1 reads_2 n sample info ## <chr> <chr> <chr> <chr> <int> <chr> <chr> ## 1 S01 CSP /storn… /storn… 10000 3D7_Dd2… 1_1 ## 2 S01 TRAP /storn… /storn… 10000 3D7_Dd2… 1_1 ## 3 S02 CSP /storn… /storn… 10000 3D7_Dd2… 1_10 ## 4 S02 TRAP /storn… /storn… 10000 3D7_Dd2… 1_10 ## 5 S03 CSP /storn… /storn… 10000 3D7_Dd2… 1_100 ## 6 S03 TRAP /storn… /storn… 10000 3D7_Dd2… 1_100 ## 7 S04 CSP /storn… /storn… 10000 3D7_Dd2… 1_500 ## 8 S04 TRAP /storn… /storn… 10000 3D7_Dd2… 1_500 ## 9 S05 CSP /storn… /storn… 10000 3D7_Dd2… 1_1000 ## 10 S05 TRAP /storn… /storn… 10000 3D7_Dd2… 1_1000
The main outputs from the demultiplex_reads function are:
• demultiplex folder: demultiplexed paired-end FASTQ files.
• demultiplex.rds: the demultiplexed table in RDS format which includes sample_id, marker_id, reads_1 (the forward read FASTQ file path), reads_2 (the reverse read FASTQ file path), n (number of demultiplexed reads), sample, info.
Before filtering and trimming the paired-end FASTQ files we can visualize the quality profiles of the forward and reverse reads of different amplicon markers by using the plot_quality function in AmpSeqR which integrates several functions from the ShortRead, DADA2, cowplot and tidyverse packages (Figure 2).
# Visualize the quality profiles of the forward and reverse reads read_quality <- plot_quality(demultiplexed) read_quality ## $CSP
## $TRAP
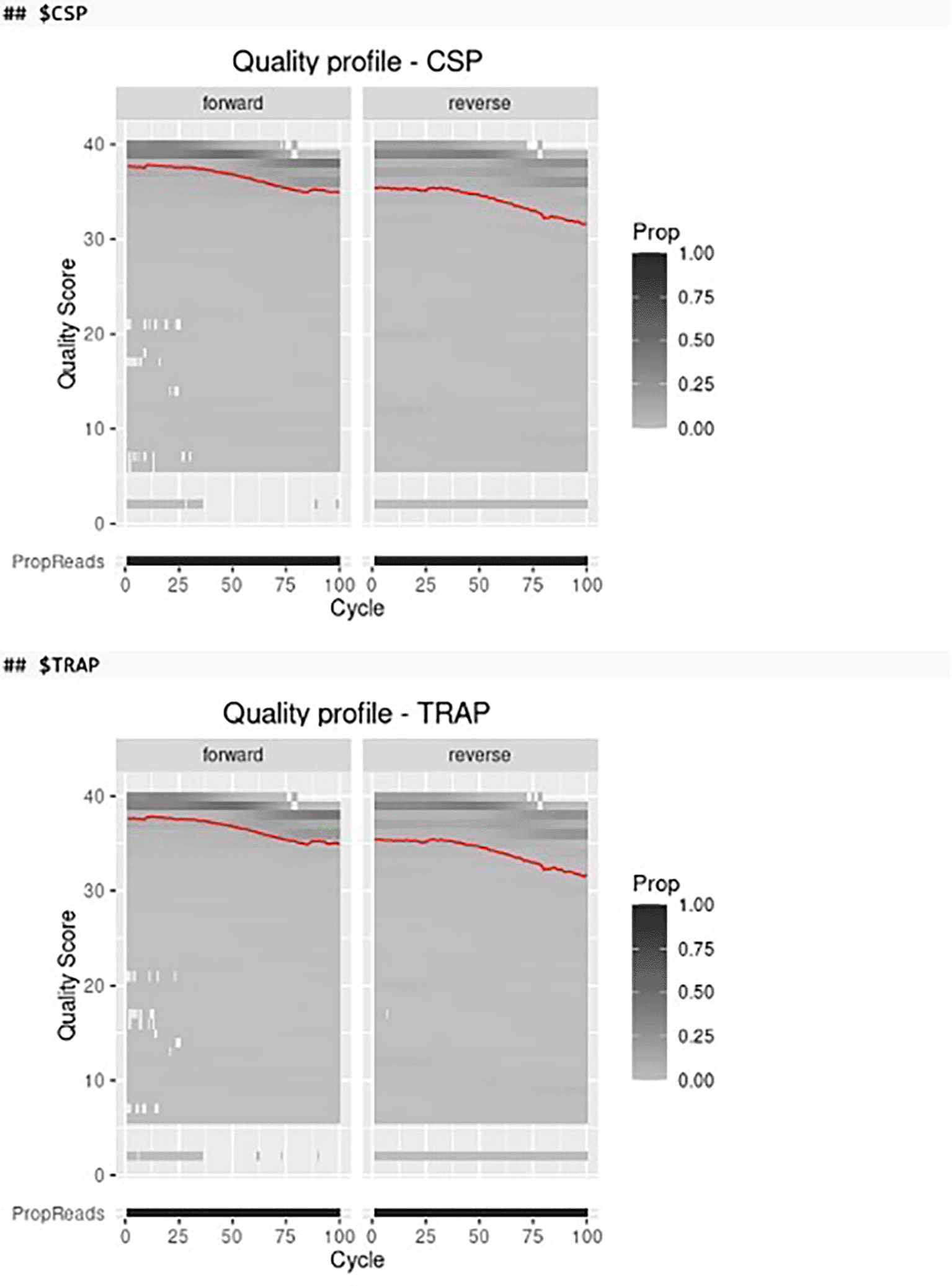
The red line shows the mean quality score of all samples in each cycle. Color represents the proportion of reads of each quality score in each cycle.
According to the quality profiles of the forward and reverse reads, we can define the trim position for the forward and reverse reads to remove low-quality bases. The marker_trim should be a data frame with three columns marker_id (character), trim_fwd (integer), and trim_rev (integer), or can be left unset (NULL, run without trimming).
The next step filters the poor-quality reads and trim low-quality bases as follows. Filtering is implemented using the dada_filter function, which uses the filterAndTrim function from the DADA2 package.
# Filter and trim reads flt_reads <- demultiplexed %>% dada_filter(output_dir = run_dir, output_sub_dir = file.path(run_dir, 'filter')) flt_reads ## # A tibble: 10 × 9 ## sample_id marker_id n sample info reads_1 reads_2 n_in n_out ## <chr> <chr> <int> <chr> <chr> <chr> <chr> <dbl> <dbl> ## 1 S01 CSP 10000 3D7_Dd2… 1_1 /storn… /storn… 10000 4878 ## 2 S01 TRAP 10000 3D7_Dd2… 1_1 /storn… /storn… 10000 4876 ## 3 S02 CSP 10000 3D7_Dd2… 1_10 /storn… /storn… 10000 4874 ## 4 S02 TRAP 10000 3D7_Dd2… 1_10 /storn… /storn… 10000 4869 ## 5 S03 CSP 10000 3D7_Dd2… 1_100 /storn… /storn… 10000 4818 ## 6 S03 TRAP 10000 3D7_Dd2… 1_100 /storn… /storn… 10000 4868 ## 7 S04 CSP 10000 3D7_Dd2… 1_500 /storn… /storn… 10000 4935 ## 8 S04 TRAP 10000 3D7_Dd2… 1_500 /storn… /storn… 10000 4933 ## 9 S05 CSP 10000 3D7_Dd2… 1_1000 /storn… /storn… 10000 4740 ## 10 S05 TRAP 10000 3D7_Dd2… 1_1000 /storn… /storn… 10000 4953
The main outputs for the dada_filter function are:
• a filter folder: filtered and trimmed paired-end FASTQ files.
• a filtered_reads.rds table: the filtered table in RDS format which includes sample_id, marker_id, n (number of demultiplexed reads), sample, info, reads_1 (the filtered and trimmed forward FASTQ file path), reads_2 (the filtered and trimmed reverse FASTQ file path), n_in (number of reads before filtering and trimming), n_out (number of reads after filtering and trimming).
The downsample_reads function randomly downsamples each sample dataset to a pre-specified number of reads (default, 10,000 reads) to reduce the computation time.
# Downsampling reads sub_reads <- flt_reads %>% downsample_reads(output_dir = run_dir, output_sub_dir = file.path(run_dir, 'downsample'), count_col = 'n_out') sub_reads ## # A tibble: 10 × 8 ## sample_id marker_id sample info reads_1 reads_2 n_out n ## <chr> <chr> <chr> <chr> <chr> <chr> <dbl> <dbl> ## 1 S01 CSP 3D7_Dd2… 1_1 /storn… /storn… 4878 4878 ## 2 S01 TRAP 3D7_Dd2… 1_1 /storn… /storn… 4876 4876 ## 3 S02 CSP 3D7_Dd2… 1_10 /storn… /storn… 4874 4874 ## 4 S02 TRAP 3D7_Dd2… 1_10 /storn… /storn… 4869 4869 ## 5 S03 CSP 3D7_Dd2… 1_100 /storn… /storn… 4818 4818 ## 6 S03 TRAP 3D7_Dd2… 1_100 /storn… /storn… 4868 4868 ## 7 S04 CSP 3D7_Dd2… 1_500 /storn… /storn… 4935 4935 ## 8 S04 TRAP 3D7_Dd2… 1_500 /storn… /storn… 4933 4933 ## 9 S05 CSP 3D7_Dd2… 1_1000 /storn… /storn… 4740 4740 ## 10 S05 TRAP 3D7_Dd2… 1_1000 /storn… /storn… 4953 4953
The main outputs for the downsample_reads function are:
• the downsample folder: downsampled paired-end FASTQ files with a read number greater than n_sample.
• the subsampled_reads.rds table: the downsampled table in RDS format includes sample_id, marker_id, sample, info, reads_1 (the downsampled forward FASTQ file path), reads_2 (the downsampled reverse FASTQ file path), n_out (number of reads before downsampling), n (number of reads after downsampling).
The dada_seq_tbl function use several functions in DADA2, Biostrings, DECIPHER, and Parallel packages, such as derepFastq, learnErrors, dada, mergePairs, AlignSeqs, DistanceMatrix to filter noise reads and merge the paired-end reads and obtain the amplicon haplotype sequence. When paired-end reads do not overlap, the min_overlap parameter should be set to -1.
# Construct amplicon sequence variant table (ASV) table seq_tbl <- sub_reads %>% dada_seq_tbl(output_dir = run_dir) seq_tbl ## # A tibble: 19 × 7 ## sample_id marker_id sequence count status sample info ## <chr> <chr> <chr> <int> <chr> <chr> <chr> ## 1 S01 CSP CCCAAATGCAAACCCAAATG… 2481 pass 3D7_D… 1_1 ## 2 S01 CSP CCCCAATGCAAACCCAAATG… 2397 pass 3D7_D… 1_1 ## 3 S01 TRAP TGGGTGAACCATGCAGTACC… 2444 pass 3D7_D… 1_1 ## 4 S01 TRAP TGGGTGAAGCATGCAGTACC… 2432 pass 3D7_D… 1_1 ## 5 S02 CSP CCCAAATGCAAACCCAAATG… 496 pass 3D7_D… 1_10 ## 6 S02 CSP CCCCAATGCAAACCCAAATG… 4378 pass 3D7_D… 1_10 ## 7 S02 TRAP TGGGTGAACCATGCAGTACC… 495 pass 3D7_D… 1_10 ## 8 S02 TRAP TGGGTGAAGCATGCAGTACC… 4374 pass 3D7_D… 1_10 ## 9 S03 CSP CCCAAATGCAAACCCAAATG… 40 pass 3D7_D… 1_100 ## 10 S03 CSP CCCCAATGCAAACCCAAATG… 4778 pass 3D7_D… 1_100 ## 11 S03 TRAP TGGGTGAACCATGCAGTACC… 63 pass 3D7_D… 1_100 ## 12 S03 TRAP TGGGTGAAGCATGCAGTACC… 4805 pass 3D7_D… 1_100 ## 13 S04 CSP CCCAAATGCAAACCCAAATG… 9 pass 3D7_D… 1_500 ## 14 S04 CSP CCCCAATGCAAACCCAAATG… 4926 pass 3D7_D… 1_500 ## 15 S04 TRAP TGGGTGAACCATGCAGTACC… 8 pass 3D7_D… 1_500 ## 16 S04 TRAP TGGGTGAAGCATGCAGTACC… 4925 pass 3D7_D… 1_500 ## 17 S05 CSP CCCCAATGCAAACCCAAATG… 4737 pass 3D7_D… 1_10… ## 18 S05 TRAP TGGGTGAACCATGCAGTACC… 5 pass 3D7_D… 1_10… ## 19 S05 TRAP TGGGTGAAGCATGCAGTACC… 4948 pass 3D7_D… 1_10…
The main outputs for the dada_seq_tbl function are:
The annotate_seq_tbl function leverages various useful functions in Biostrings and DECIPHER to determine the amplicon haplotype status and to distinguish the true haplotype sequences from sequencing background noise and artifacts, including ultra-rare variants and chimeric reads generated during PCR amplification.
# Haplotype inference seq_ann_tbl <- seq_tbl %>% annotate_seq_tbl(marker_info = marker_info, output_dir = run_dir) seq_ann_tbl ## # A tibble: 19 × 9 ## sample_id marker_id sequence count status sample info ident ident_z ## <chr> <chr> <chr> <int> <chr> <chr> <chr> <dbl> <dbl> ## 1 S01 CSP CCCAAATG… 2481 pass 3D7_D… 1_1 1 1.05 ## 2 S01 CSP CCCCAATG… 2397 pass 3D7_D… 1_1 0.892 -0.843 ## 3 S01 TRAP TGGGTGAA… 2444 pass 3D7_D… 1_1 1 0.949 ## 4 S01 TRAP TGGGTGAA… 2432 pass 3D7_D… 1_1 0.958 -0.949 ## 5 S02 CSP CCCAAATG… 496 pass 3D7_D… 1_10 1 1.05 ## 6 S02 CSP CCCCAATG… 4378 pass 3D7_D… 1_10 0.892 -0.843 ## 7 S02 TRAP TGGGTGAA… 495 pass 3D7_D… 1_10 1 0.949 ## 8 S02 TRAP TGGGTGAA… 4374 pass 3D7_D… 1_10 0.958 -0.949 ## 9 S03 CSP CCCAAATG… 40 pass 3D7_D… 1_100 1 1.05 ## 10 S03 CSP CCCCAATG… 4778 pass 3D7_D… 1_100 0.892 -0.843 ## 11 S03 TRAP TGGGTGAA… 63 pass 3D7_D… 1_100 1 0.949 ## 12 S03 TRAP TGGGTGAA… 4805 pass 3D7_D… 1_100 0.958 -0.949 ## 13 S04 CSP CCCAAATG… 9 pass 3D7_D… 1_500 1 1.05 ## 14 S04 CSP CCCCAATG… 4926 pass 3D7_D… 1_500 0.892 -0.843 ## 15 S04 TRAP TGGGTGAA… 8 pass 3D7_D… 1_500 1 0.949 ## 16 S04 TRAP TGGGTGAA… 4925 pass 3D7_D… 1_500 0.958 -0.949 ## 17 S05 CSP CCCCAATG… 4737 pass 3D7_D… 1_10… 0.892 -0.843 ## 18 S05 TRAP TGGGTGAA… 5 pass 3D7_D… 1_10… 1 0.949 ## 19 S05 TRAP TGGGTGAA… 4948 pass 3D7_D… 1_10… 0.958 -0.949
The main outputs for the annotate_seq_tbl function are:
• the seq_ann_tbl.rds table: the annotated amplicon haplotype sequence table in RDS format includes sample_id, marker_id, sequence (haplotype sequence), count (read counts), status (inferred haplotype status), sample, info, ident (sequence similarity), ident_z (standardized sequence similarity).
The amplicon sequence haplotype status includes the following states:
• pass: the amplicon sequences haplotype has passed all quality control checks,
• low_sample_count, failed: the sample read count lower than the minimum number of read per sample per marker,
• low_asv_count, failed: the amplicon sequence haplotype counts lower than the minimum number of read for each haplotype,
• low_asv_freq, failed: the amplicon sequence haplotype frequency lower than the minimum haplotype frequency,
• low_ident, failed: the amplicon sequence haplotype similarity (after being mapped to the reference) is lower than the minimum sequence similarity,
• low_ident_z, failed: the standardized amplicon sequence haplotype similarity (after being mapped to the reference) lower than the minimum standardized sequence similarity,
• chimera, failed: the sequence is a chimera of reads,
• multiple, failed: multiple fail types.
The sequence_filter function is mainly optimizing amplicon haplotype calling and uses several parameters, such as sample missingness, amplicon marker missingness, the number of alleles at a locus, as well as variant heterozygosity and MAF in a given dataset. This function is implemented with Biostrings, magrittr, withr, GenomicRanges, VariantAnnotation, S4Vectors, IRanges, and tidyverse R packages.
# Haplotype filtering seq_flt_tbl <- sequence_filter(seq_ann_tbl = seq_ann_tbl, sample_manifest = sample_manifest, marker_info = marker_info, output_dir = run_dir, vcf_output_dir = file.path(run_dir, 'vcf'), sample_med_He = 0.1 ) seq_flt_tbl ## # A tibble: 19 × 11 ## sample_id marker_id sequence count masked status ident haplo…1 frequ…2 sample ## <chr> <chr> <chr> <int> <lgl> <chr> <dbl> <chr> <dbl> <chr> ## 1 S01 CSP CCCAAAT… 2481 FALSE pass 1 CSP-2 0.509 3D7_D… ## 2 S01 CSP CCCCAAT… 2397 FALSE pass 0.892 CSP-1 0.491 3D7_D… ## 3 S01 TRAP TGGGTGA… 2444 FALSE pass 1 TRAP-1 0.501 3D7_D… ## 4 S01 TRAP TGGGTGA… 2432 FALSE pass 0.958 TRAP-2 0.499 3D7_D… ## 5 S02 CSP CCCAAAT… 496 FALSE pass 1 CSP-2 0.102 3D7_D… ## 6 S02 CSP CCCCAAT… 4378 FALSE pass 0.892 CSP-1 0.898 3D7_D… ## 7 S02 TRAP TGGGTGA… 495 FALSE pass 1 TRAP-1 0.102 3D7_D… ## 8 S02 TRAP TGGGTGA… 4374 FALSE pass 0.958 TRAP-2 0.898 3D7_D… ## 9 S03 CSP CCCAAAT… 40 FALSE pass 1 CSP-2 0.0083 3D7_D… ## 10 S03 CSP CCCCAAT… 4778 FALSE pass 0.892 CSP-1 0.992 3D7_D… ## 11 S03 TRAP TGGGTGA… 63 FALSE pass 1 TRAP-1 0.0129 3D7_D… ## 12 S03 TRAP TGGGTGA… 4805 FALSE pass 0.958 TRAP-2 0.987 3D7_D… ## 13 S04 CSP CCCAAAT… 9 FALSE pass 1 CSP-2 0.00182 3D7_D… ## 14 S04 CSP CCCCAAT… 4926 FALSE pass 0.892 CSP-1 0.998 3D7_D… ## 15 S04 TRAP TGGGTGA… 8 FALSE pass 1 TRAP-1 0.00162 3D7_D… ## 16 S04 TRAP TGGGTGA… 4925 FALSE pass 0.958 TRAP-2 0.998 3D7_D… ## 17 S05 CSP CCCCAAT… 4737 FALSE pass 0.892 CSP-1 1 3D7_D… ## 18 S05 TRAP TGGGTGA… 5 FALSE pass 1 TRAP-1 0.00101 3D7_D… ## 19 S05 TRAP TGGGTGA… 4948 FALSE pass 0.958 TRAP-2 0.999 3D7_D… ## #… with 1 more variable: info <chr>, and abbreviated variable names ## # 1haplotype, 2frequency
The main outputs for the sequence_filter function are:
• the seq_flt_tbl.rds table: the final amplicon haplotype sequence table in RDS format which includes sample_id, marker_id, sequence (haplotype sequence), count (read counts), masked (TRUE indicates the sequence contains variants that might be errors, and replaces the nucleotide with the reference genome nucleotide), status (haplotype status, all pass), ident (sequence similarity), haplotype (a named panel of haplotypes for each marker), frequency (within-sample haplotype frequency for each marker), sample, info.
The generate_report function generates a HTML report including various summary data visualizations. The function is based on Biostrings, Rmarkdown, plyr, DT, plotly, viridisLite, stats, flextable, ComplexHeatmap, scales, heatmaply, digest, tidyselect, data.table, DECIPHER, htmltools, pheatmap, ape, randomcoloR, ggtree, ggstance, and tidyverse R packages. The HTML summary report for the synthetic P. falciparum dataset is available from Figshare (https://doi.org/10.6084/m9.figshare.21739121).27
# Data visualization generate_report(sample_manifest = sample_manifest, marker_info = marker_info, demultiplexed = demultiplexed, flt_reads = flt_reads, sub_reads = sub_reads, seq_ann_tbl = seq_ann_tbl, seq_flt_tbl = seq_flt_tbl, report_output_dir = file.path(run_dir, 'report')) ## ---- generating report ---- ## report save to "runs/report/Report.html"
We also include the process_run function that calls all the functions, making it easy to get all results.
# Create the analysis directory process_run_dir <- "runs_process" dir.create("./runs_process") # Input data reads_1 <- example_data$reads_1 reads_2 <- example_data$reads_2 sample_manifest <- example_data$sample_manifest marker_info <- example_data$marker_info # Fast run process_run(reads_1 = reads_1, reads_2 = reads_2, sample_manifest = sample_manifest, marker_info = marker_info, run_dir = process_run_dir, min_marker_count = 500, sample_med_He = 0.1) ## ---- processing run ---- ## ---- demultiplexing ---- ## ---- filter and trim ---- ## ---- downsampling ---- ## ---- ASV estimation ---- ## ---- filtering haplotype sequence ---- ## ---- generating report ----
For the SARS-CoV-2 AmpSeq data, the paired-end FASTQ data is already demultiplexed by sample. We first demultiplexed by marker and then run AmpSeqR from the filter and trim step as follows. Some output from the HTML report for the SARS-CoV-2 dataset is shown in Figure 3.
# Input data sample_manifest <- read_csv("Covid19/sample_manifest.csv", col_types = cols()) marker_info <- read_csv("Covid19/marker_info.csv", col_types = cols(start = col_integer())) # The paired-end FASTQ data already demultiplexed demultiplexed <- readRDS("Covid19/demultiplex.rds") # Create the analysis directory dir.create("./runs") # Filter and trim reads flt_reads <- demultiplexed %>% dada_filter(output_dir = run_dir, output_sub_dir = file.path(run_dir, 'filter')) # Downsampling reads sub_reads <- flt_reads %>% downsample_reads(output_dir = run_dir, output_sub_dir = file.path(run_dir, 'downsample'), min_read_count = 0, count_col = 'n_out') # Construct amplicon sequence variant table (ASV) table seq_tbl <- sub_reads %>% dada_seq_tbl(output_dir = run_dir) # Haplotype inference seq_ann_tbl <- seq_tbl %>% annotate_seq_tbl(marker_info = marker_info, output_dir = run_dir, min_marker_count = 0, min_asv_count = 0) # Haplotype filtering seq_flt_tbl <- sequence_filter(seq_ann_tbl = seq_ann_tbl, sample_manifest = sample_manifest, marker_info = marker_info, output_dir = run_dir, vcf_output_dir = file.path(run_dir, 'vcf'), max_sm_miss = 1, max_marker_miss = 1, min_homo_rep = NULL, terminal_region_len = NULL ) # Data visualization generate_report(sample_manifest = sample_manifest, marker_info = marker_info, demultiplexed = demultiplexed, flt_reads = flt_reads, sub_reads = sub_reads, seq_ann_tbl = seq_ann_tbl, seq_flt_tbl = seq_flt_tbl, report_output_dir = file.path(run_dir, 'report'))
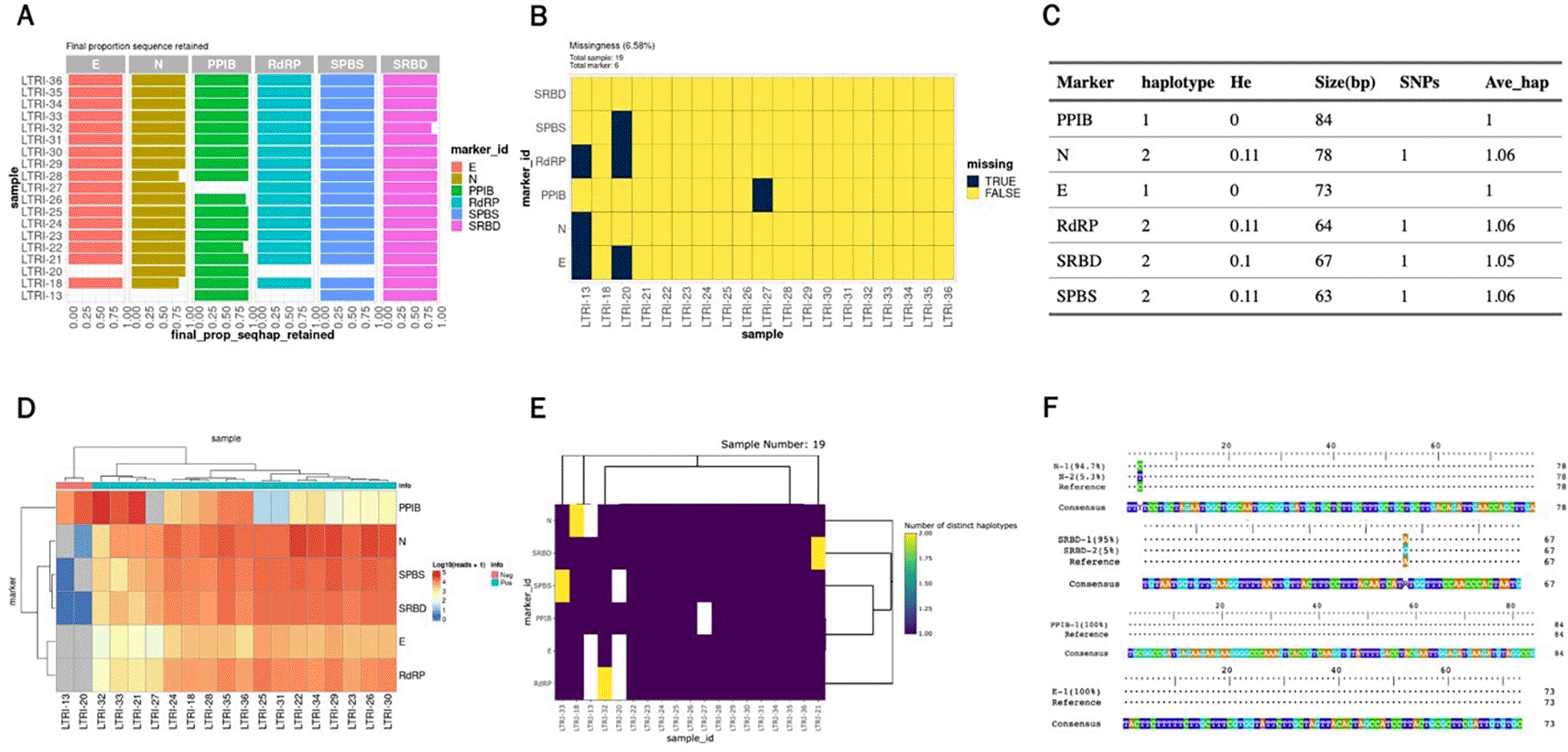
A. The proportion of sequences that are finally retained. B. Missing values in the dataset. C. Haplotype diversity metrics for amplicon markers in the dataset. D. Read counts for amplicon markers and all samples in the dataset. E. Number of unique haplotypes detected per amplicon per sample. F. Visualisation of haplotype and reference sequences.
The detectability of the minor clone of the CPMP and CSP amplicon markers under different mixture ratios of P. falciparum is shown in Figure 4. The correct minor haplotype was detected in mixture proportions from 1:1 to 1:3000 in most mixtures and detected haplotype frequencies similar to the defined mixture proportion. These results demonstrate that AmpSeqR can detect low-frequency haplotypes accurately.

The x-axis represents the haplotype frequency, and the y-axis represents the sample. Colored by haplotype and labelled by haplotype and haplotype frequency. The grey bar represents a reference for different defined mixture proportions.
We present the AmpSeqR R package for analysis of AmpSeq data which was primarily developed for the analysis of infectious diseases. The pipeline offers various useful steps including data pre-processing, amplicon sequence variants (ASVs) estimation, data post-processing, data visualization and includes several parameters to filter noise reads and improve the accuracy of the detected haplotype. AmpSeqR allows users to easily analyze the AmpSeq data and generate an HTML report including various summary data visualizations. Additionally, the one-line commands for processing the data do not require extensive R knowledge which provides a user-friendly experience for R users of all levels of experience.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
1. Since AmpSeq is being used in Plant Pathology, I would have liked to see an example ... Continue reading Nice paper, with detailed pipeline for the analysis. However, I have some comments.
1. Since AmpSeq is being used in Plant Pathology, I would have liked to see an example using AmpSeqR for plant studies.
2. The computation time to justify why it is better than already existing ones and the system requirement (PC).
1. Since AmpSeq is being used in Plant Pathology, I would have liked to see an example using AmpSeqR for plant studies.
2. The computation time to justify why it is better than already existing ones and the system requirement (PC).