Keywords
human cancers, vitamin E, tocotrienols, anticancer mechanisms, Vitamin E, C, D, Genomics, Biomarkers
This article is included in the Oncology gateway.
human cancers, vitamin E, tocotrienols, anticancer mechanisms, Vitamin E, C, D, Genomics, Biomarkers
The revised version of the manuscript incorporates the corrections suggested by the reviewers. The Introduction and methodology were revised as suggested. Search strategy used and the inclusion and exclusion criteria were explained more clearly. It needs to be emphasised that this is a scoping review and the analysis is based on secondary data only, that were analysed using an online bioinformatic software. Reviewer had pointed out that the record numbers in Fig 1 did not tally, and we have checked our data and found an error and we have rectified this. In addition, Figure 2 has been revised to include information on the number of relevant papers published from 2015 to 2021 as well as the forms of tocotrienols used in these papers. Table 1 have been revised to improve clarity of data presented. Finally, an extensive grammar check was performed, which improving the clarity and coherence of the manuscript.
See the authors' detailed response to the review by Kok Lun Pang
Cancers are the second leading cause of death worldwide before age seventy.1 Cancers may be formed due to gene mutations or critical dysregulation of different cellular survival and death mechanisms,2 usually caused by injurious agents that affect cellular homeostasis, structures, and organelles. Some of the more lethal cellular disruptions are associated with an injury to the plasma membrane, the mitochondria, the endoplasmic membrane and the nucleus,3 which can lead to the development of malignancies. Regardless of the advanced management and treatment available, the number of cancer cases reported is steadily increasing.1 Treating cancers can be complicated as treatments are generally tailored to each patient's needs and health circumstances, including the cancer origin, position, lymph node involvement and metastasis. Cancer treatments are broadly categorized based on the main method of treatment, such as surgical resection, chemotherapy, radiotherapy and hormonal therapy.4 Most of the therapeutic approaches currently used have well documented complications. For instance, the surgical approach requires hospitalization and wound healing time, including the possibility of experiencing common post-surgical complications. Chemotherapeutics drugs are cytotoxic to most living cells (normal or malignant), causing side effects such as hair loss, fatigue, nausea, different vital organs toxicity and potentially, the of developing drug resistance. Hormonal therapies, on the other hand, increase susceptibility to other cancers along with other complications such as gynecomastia and hirsutism. Hence, the need for newer ways to treat and/or prevent cancer which do not pose as many side effects as observed with current therapies or have the ability to decrease the severity of these side effects. Hence, there is a need to look for alternative compounds that are effective and have lesser side effects, such as plant phytochemicals.
The evidence in the literature suggests that several natural compounds such as plant phytochemicals and vitamins are rapidly emerging as promising anticancer agents, where these compounds have been used as therapeutic agents or adjuvants to treat cancers,5 promote immunity, protect against DNA damage, and regulate cell cycle and proliferation.5 Examples of such phytochemicals include isoprenoid derivatives, phenol compounds, carbohydrate derivatives, fatty acids, amino acids, and minerals. This scoping review will focus on the anticancer activities of tocotrienols (T3), a naturally occurring form of vitamin E.
Vitamin E (Vit E) is a naturally occurring plant phytochemical that exist naturally as two isoprenoid derivatives, tocotrienols (T3) and tocopherols (TP). Tocotrienols and TP have similar chemical structures. However, the side chain of TP is saturated, while the side chain of T3 contains three unsaturated bonds. In addition, both TPs and T3 can be found in four distinctive isoforms known as alpha (α), beta (β), delta (δ) and gamma (γ), which are classified based on the position of methyl groups on the chromanol ring of vitamin E. Natural sources of T3 include palm oil,6–8 annatto seeds9 and rice bran,10,11 while TP can be found in olive oil, soy oil and corn oil.
According to the literature, T3 possess potent antioxidant, cardioprotective, neuroprotective and anticancer activities, reported in many in vitro and in vivo studies.12 Tocotrienols exert anticancer effects mainly through differentially regulating molecular targets responsible for programmed cell death (PCD) signals, angiogenesis and metastasis.13 The anticancer activity of T3 have been demonstrated in several types of cancers, including breast cancer (BC),14,15 pancreatic cancer,16,17 hepatic cancer,18,19 haematological cancer20,21 and melanoma.22,23 The cytotoxic effects of T3 against cancer cells are triggered via activation of various intracellular signalling pathways such as activation of apoptosis (programmed cell death type I), autophagy (programmed cell death type II), regulation of cell cycle through inhibiting cell cycle progression through cyclin kinase-dependent (CDK), and cyclin D1, and suppression of angiogenesis. Furthermore, T3, may synergize with chemotherapeutic drugs such as statins,24 celecoxib25 and tamoxifen,26,27 to augment their efficacy. Combinations of T3 and natural dietary compounds such as ferulic acid (FA) or jerantinine was reported to induce higher anticancer effects.28,29 The cytotoxic effects of T3 on cancer cells suggest that these molecules can potentially be used as an effective tool in cancer treatment.
Although there is growing evidence to support the anticancer activity of T3, there is a need to understand the cellular actions and the regulatory pathways on how T3 induce anticancer effects in human cancer cells as different mechanisms have been reported in different human cancer cell lines. Hence, the aim of this scoping review is to understand the cellular actions and the regulatory pathways on how tocotrienols induce anticancer effects in human cancer cells through analysing the genes and proteins that are commonly affected in these cancer cells following exposure to T3 in research articles published between January 2015 to September 2021.
This review was carried out according to the guidelines of the Preferred Reporting Items For Systematic Review And Meta-Analysis (PRISMA).30 Six databases (PubMed, Pro-Quest, Web of Science, Scopus, Ovid Medline and Embase) were searched using Medical Subject Heading (MeSh) and keywords “tumor*”, “gene*”, “protein*”, “tocotrienol*”, “neoplasm”, “cancer,” and “cell line” and Boolean commands such as “tumor* OR neoplasm Or cancer” AND “gene(s)” OR “gene* expression” OR “protein(s)” AND “tocotrienol*”. Papers reporting on the effects of T3 on various types of human cancer cell lines, with the primary outcome being gene modulation, regulation or amplification and protein dysregulation, were selected for analysis. In addition, the search was restricted to papers published in English in the last seven years (January 2015 to September 2021) to use the latest evidence for this study.
Eligible studies were selected based on the study’s inclusion and exclusion criteria. The inclusion criteria include that the paper must (i) be an original research article reporting on human cancer cell line exposed to T3 and the findings were compared with untreated cells; (ii) reported on the effects of T3 using differential expression genes or proteins. The exclusion criteria were (i) studies, where cancer cells were not directly exposed to T3; (ii) the effects of T3 on the human cancer cells did not report on gene or protein regulation; (iii) in vivo studies; (iv) in silico studies. Any papers that did not meet the inclusion criteria will be excluded from this scoping review.
Two step screening approach was used to identify suitable articles. Original articles published up to 30 Sept 2021 were included in the analysis. The articles identified from the six databases (PubMed, Pro-Quest, Web of Science, Scopus, Ovid Medline & Embase) were imported and compiled in Endnote software31 initially to eliminate any duplicate articles, and then the articles were uploaded into Covidence software,32 an online software used to carry out systematic reviews. The first screening step was the title and abstract review and this was followed by a second screening step, which was full text screening and finally data extraction. In each screening step, two researchers (SMM and US) independently performed the article search, screening and data extraction and conflicts were resolved by a third independent researcher (AKR). Then, data extraction was performed using an Excel template. The following data were extracted from each article: origin and type of cancer cell line, dose, duration and type of tocotrienol or tocotrienol isomers the cancer cells were exposed to, gene and protein modulation, regulation or expression as the primary outcome and other outcomes such as (apoptosis, cell cycle arrest, IC50), and, the bibliographic details of the article (e.g., authors, year of publication, journal name).
The search yielded a total of 619 records from six databases [ProQuest (n= 61), PubMed (n= 84), Embase (n = 148), Ovid Medline (n =53), Scopus (n = 137), Web of Science (n =136)]. The articles were imported into Endnote, a referencing software and after removal of duplicate articles (n=145) from the six databases, there were 474 articles left (Figure 1). These 474 records were uploaded to Covidence, an online software (www.covidence.com) that enables researchers to independently screen and review articles. In Covidence, another 151 duplicate articles were identified and removed from the pool, which left 323 articles in the pool. From the title and abstract screening, based on the inclusion and exclusion criteria, a total of 252 articles were excluded. The remaining 96 articles were subjected to full text review, where 59 articles were excluded, leaving a total of 37 studies (Table 1) that were eligible for data extraction (Figure 1). Data were extracted using a MS-Excel sheet, and information such as origin and type of cancer cell line, dose, duration and type of T3 used and year of publication were collected (Table 1).

| Author, Year, Ref | Human cancer cell line | Cancer type | T3 | Intervention | Genes/Proteins modulated | Main outcome | Other outcomes (e.g., apoptosis, cell cycle, IC50) |
|---|---|---|---|---|---|---|---|
| Sazli et al., (2015)47 | HepG2 | Hepato-cellular carcinoma | γT3 | 70 μM (48 hrs) | PRX4 | ↓ | |
| Chen et al., (2015)50 | HL-60 | Acute myeloid leukaemia | γT3 | 10, 20 or 30 μM (12 hrs) | GLO1, HMGCR, N-RAS, H-RAS, K-RAS, RAF-1, p-AKT, p-ERK | ↓ | |
| Gu et al., (2015)33 | CSC from MCF-7 | ER+ BC | γT3 | 1-5 μg/mL (7-8 days) | SHP2, P- SHP2, H-RAS, K-RAS, p-ERK | ↓ | |
| CSC from Hela | Cervical adeno-carcinoma | ||||||
| Ryosuke et al., (2015)9 | PC3 | Androgen--independent prostate cancer | Mixed T3 (90% δT3 and 10% γT3) | 10, 20 μg/mL (24 hrs) | p-SRC, p-STAT3 | ↓ | |
| Parajuli et al., (2015)34 | MCF-7 | ER+ BC | γT3 | 0-8 mM (96 hrs) | P27 (CDKN1B) | ↑ | |
| c-MYC, p-RB, E2F1, CDK4, cyclin D1 (CCND1), FBW7, p-GSK3 α/β, p-AKT, PI3K, p-MTOR, p-ERK 1/2, p-MEK 1/2 | ↓ | ||||||
| Shibata et al., (2015)55 | DLD-1 | CRC | δT3 | 0–20 μM (12, 24 or 48 hrs) under hypoxia/normoxia conditions) | P21 (CDKN1A), P27 (CDKN1B), CASP3, CASP9 | ↑ | |
| HIF-1α, AKT, CDK4 | ↓ | ||||||
| Wang et al., (2015)63 | MiaPaCa-2 | Pancreatic cancer | δT3 | 50 μM (12 hrs) | EGR-1, BAX, SLC20A1, SPRY4, ARRDC3, JUN CFLAR, CAPZA2 | ↑ | |
| RGS22, SERPINA3, DOCK11, GJA7, GBP2, SLCO2A1, CCL14 | ↓ | ||||||
| Tiwari et al., (2015)37 | MCF7 | ER+ BC | γT3 | 40 μmol/L (6,12 & 24 hrs) | LC3B-I, LC3B-II, BEC-1, ATG5, ATG12, LAMP-1, Cathepsin-D, BAX, CASP3, PARP, BIP, IRE1, p-ERK, p-elf2α, ATF-4, CHOP, TRB3, P38, p-JNK 1/2 | ↑ (both cell lines) | |
| MDA-MB-231 | TNBC | ||||||
| BCL2, p-ERK 1/2 | ↓ (both cell lines) | ||||||
| Wang et al., (2015)38 | MDA-MB-231 | TNBC | δT3 | 0,10, 30, 100 μM (24 hrs) | miR-429 | ↑ | |
| MDA-MB-468 | |||||||
| Burdeos et al., (2016)48 | HepG2 | Hepatocellular Carcinoma | γT3, δ T3 | 5, 10,15, 20 μM (12-24 hrs) | GRB2, SOS1, cellular- SRC (c-SRC), SHC2, HRAS, p-SHC | ↓ | |
| Comitato et al., (2016)40 | Hela | Cervical adenocarcinoma | TRF, | 10 μg/mL | p-IRE-1α | ↑ (δ T3) in Hela cells | |
| MCF7 | ER+ BC | αT3 | 5 μg/mL | CASP12& CASP8 | ↑ (δ T3, TRF, γT3) in Hela cells | ||
| γT3 | 5, 10, 20 μg/mL | CASP9 | ↑ (δ T3) in Hela cells | ||||
| δT3 | 5 &10 μg/mL | REBF1, SCD & LPIN1 | ↓ (α T3, δ T3, γT3) | ||||
| HSPA5, ASNS, PHLDA1, GDF15& sXBP1 | ↑ (δ T3, γT3) | ||||||
| SREBF2, CDKN1A& ID2, | ↓ (δ T3, γT3) | ||||||
| CND1, CHAC1, DNAJB9, FAS, GEM, GFPT1& XBP-1 | ↑ (γT3) | ||||||
| GSK3B, DNAJC10& c-JUN | ↓ (δ T3) | ||||||
| DF2L1, BCR, TRIB3& FAM129A | ↑ (δ T3) | ||||||
| Eitsuka et al., (2016)56 | DLD-1 | CRC | δ T3 | 10-12.5 μM (72 hrs) | Human TERT (hTERT) mRNA | ↓ | |
| FA | 20 μM (72 hrs) | hTERT | ↓ | ||||
| Khallouki et al., (2016)35 | MCF-7 | ER+ BC | δ-T3 | 500 μM (16 hrs) | TGFα, PR, Ps2 | ↑ | |
| HELN cells (HELA cells transfected ERα or Erβ) | Cervical cancer | ||||||
| Montagnani et al., (2016)52 | BLM (NRAS mutated) | Melanoma | δ-T3 | 20 μg/mL (18 hrs) | c- CASP3, IRE1α, ATF4, BIP, p-EIF2α, p-ERK, RE1α, PDI, BAX, BCL2, PARP, CASP4 | ↑ (both cell line) | |
| A375 (V600E BRAF mutated) | |||||||
| ERO1α | ↑ A375 cells | ||||||
| Abubakar et al. (2016)57 | U87MG | GBM | δ-T3 | 3.12 ± 1.26 μg/mL (72hrs) | CASP3 | ↑ (both cell line) | |
| HT29 | CRC | 5.71 ± 1.23 μg/mL (72hrs) | CASP8 | ↑ (both cell line) | |||
| Prasad et al., (2016)60 | HCT 116 (mutated K-ras) | CRC | γT3 | 10,25,30 μM (24 hrs) | CCND1, MMP9, SURVIVIN, c-IAP-1, c-IAP-2, cellular MYC (cMYC), CXCR4, VEGF, NF-KB | ↓ | |
| HT-29 | |||||||
| Caco-2 (wild type K-ras) | |||||||
| Huang et al., (2017)11 | VCaP cells | Prostate cancer | αT3 | 20 μM (24 hrs) | P21 (CDKN1), P27 (CDKN1) | ↓ | |
| γT3 | 20 μM (24 hrs) | Cyclin D1 (CCND1), Cyclin A (CCNA) | ↑ | ||||
| δ-T3 | 10 μM (24 hrs) | ||||||
| Lee et al., (2017)51 | K562 | Chronic myeloid leukaemia | δ-T3 | 8.04 μg/mL (72 hr) | BAD, BCL2L1, BNIP, HRK, MCL1, APAF1, BIRC3, NOD1, NOL3, PYCARD, CASP7, CASP8, CASP9, GADD45A, TP53, TP73, CD40, FAS, TNFERSF10B, TNFERSF1A,TNFERSF9, CD70, FASLG& DAPK1 | ↑ | |
| Rajasinge et al., (2017)10 | H520 | NSCLC squamous cell carcinoma | TRF | 0.04, 0.08 and 0.12 mg/mL (48 hrs) | NOTCH-1, SURVIVIN, PARP, HES-1, BCL-XL | ↓ (both cell lines) | |
| A549 | NSCLC adenocarcinoma | ||||||
| Xu et al., (2017)54 | HeLa cells | Cervical cancer | γT3 | 30,45 & 60 μM (12-24 hrs) | BAX, C-PARP, CASP3,C-CASP9, Cytochrome c (mitochondria) | ↑ | |
| BCL2, Ki67, PCNA | ↓ | ||||||
| Zappe et al., (2018)58 | Caco-2 | CRC | αToc | 20 IU/mL (48 hrs) | MLH1, DNMT1 | ↑ | |
| T3 | 5 mg/mL (48 hrs) | ||||||
| T3 | 2 mg/mL (48 hours) | ||||||
| Kaneko et al., (2018)41 | PC3 | Prostate cancer | δT3 | 0-40 μM (12 hours under hypoxic conditions) | HIF-1α, HIF-2α | ↓ | |
| Rajasinghe et al., (2018)45 | A549 | Lung cancer | δT3 | 10, 20, 30 μM (72 hours) | miR-451 | ↑ | |
| H1299 | NSCLC | MMP9, NOTCH-1, UPA, HES-1 | ↓ | ||||
| Zhang et al., (2018)62 | MGC-803 | Gastric adenocarcinoma | γT3 | 0, 15, 30, 45 & 60 μmol/L (24,48 & 72 hrs) | MMP2, MMP9 | ↓ | |
| SGC-7901 | |||||||
| Palau et al., (2018)16 | MIA PaCa-2 (K-Ras mutated) | Pancreatic carcinoma | γT3 | 40 μM (2,4,6 hrs) | SPT, DEGS1, CERS6l, ASM, ARV1 | ↑ (all cell lines) | |
| Panc 1 (wild type) | CERT | ↓ (all cell lines) | |||||
| BxPc3 (K-Ras mutated) | |||||||
| Dronamraju et al., (2019)36 | MCF-7 | ER+ BC | γT3 | 4-7 μM (4 days) | p-AMPKα, p-AMPKß | ↑ (both cells) | |
| MDA-MB-231 | TNBC | HKII, PFKP, PK-M2, LDH, LKB1, p-FOXO3a | ↓ both cell) | ||||
| AKT, FOXO3, MTOR, PFKFB2& PRKAG1 | ↓ (MCF7) | ||||||
| AKT2, FOXO3, MTOR, PFKFB2 SLC2A4 & STK11 | ↓ (MDA-MB-231) | ||||||
| MLYCD, PRKAG1, PRKAG2 & STRADA | ↑ (MDA-MB-231) | ||||||
| Husain et al., (2019)59 | HCT-116 | CRC | δT3 | 50 μMol (24 hrs) | E-cadherin, C-PARP | ↑ (both cells) | |
| SW620 | β-catenin, Vimentin, NF-kB/P65, MMP9, VEGF | ↓ both cell) | |||||
| Sun et al., (2019)61 | SGC-7901 | Gastric cancer | γ-T3 | 15, 30, 45, or 60 μmol/L (/24 hrs) | PP2A, p-ATM | ↑ | |
| NF-kB/p65 | ↓ | ||||||
| Tang et al., (2019)43 | PC3 cells | Prostate cancer (androgen-independent) | γT3 | 10 μg/mL (72 for under serum-free condition) | ANG-1/TIE-2 | ↓ (combined with Tie-2 inhibitor) | |
| ANG-1 | ↓ | ||||||
| Ramdas et al. (2019)39 | MDA-MB-231 | TNBC | γT3 | 5.8 μg/mL (24 hrs) | >30 proteins | ↑ | |
| >30 proteins | ↓ | ||||||
| Moore et al., (2020)44 | LNCaP | Prostate cancer (androgen-sensitive) | γT3 | 10 to 80 μM (6 hrs) | ERK, p-c-JUN, Casp3, Casp9 | ↑ (both cell lines) | |
| PC-3 | Prostate cancer (androgen-independent) | ||||||
| Tupal et al., (2020)49 | HUH-7 | Hepatocellular carcinoma | αT3 | 15 ± 0.4 μM (24 hrs) | BAX, BID | ↑ (αT3 lower than αT3-loaded NLC) | |
| αT3-loaded NLC | 0.6 μM (24 hrs) | mcl-1 mRNA, SURVIVIN, Cyclin-B1 (CCNB1) | ↓ (αT3 lower than αT3-loaded NLC) | ||||
| Fontana, et al., (2020)42 | PC3 | Prostate cancer (androgen-dependent) | δT3 | 15 μg/mL (1-24 hrs) | c-CASP 3, JNK, P38 | ↑ (both cell lines) | |
| DU145 | OXPHOS, c-LOPA1 | ↓ (both cell lines) | |||||
| p-AKT, PINK, PARKIN, P62/SQSTM1 | ↑ (PC3 cells) | ||||||
| p-AKT, p-AMPK, MFN2, AKT/MTOR | ↓ (PC3 cells) | ||||||
| Idriss et al., (2020)14 | MDA-MB-231 | TNBC | βT3 | 10, 20, 30, 40, 50 μM for 24 to 48 hrs | Cytochrome c, c-CASP3, c-PARP1 | ↑ | |
| BCL2, p-PI3K, p-GSK3 | ↓ | ||||||
| Ding et al., (2021)26 | MCF-7/Adr (multidrug resistant) | ER+ BC | γT3 | 25 or 50 μM) for 72 hrs | MDR1 | ↑ | |
| MDR1/P-GP, P-GP | ↓ | ||||||
| Shen et al., (2021)46 | CNE1 | NPC | δT3 | 10, 20, 40, 80 μM (24 hrs) | >30 genes | ↓ | |
| >30 genes | ↑ | ||||||
| Raimondi et al. (2021)53 | A375 BLM | Melanoma | δT3 | 15 μg/mL (6,12 or 24 hrs) | AMPK, p-JNK, p-P38, p-ERK1/2 | ↑ (both cell lines) | Trigger paraptosis in melanoma cells via the Ca2+/ROS axis |
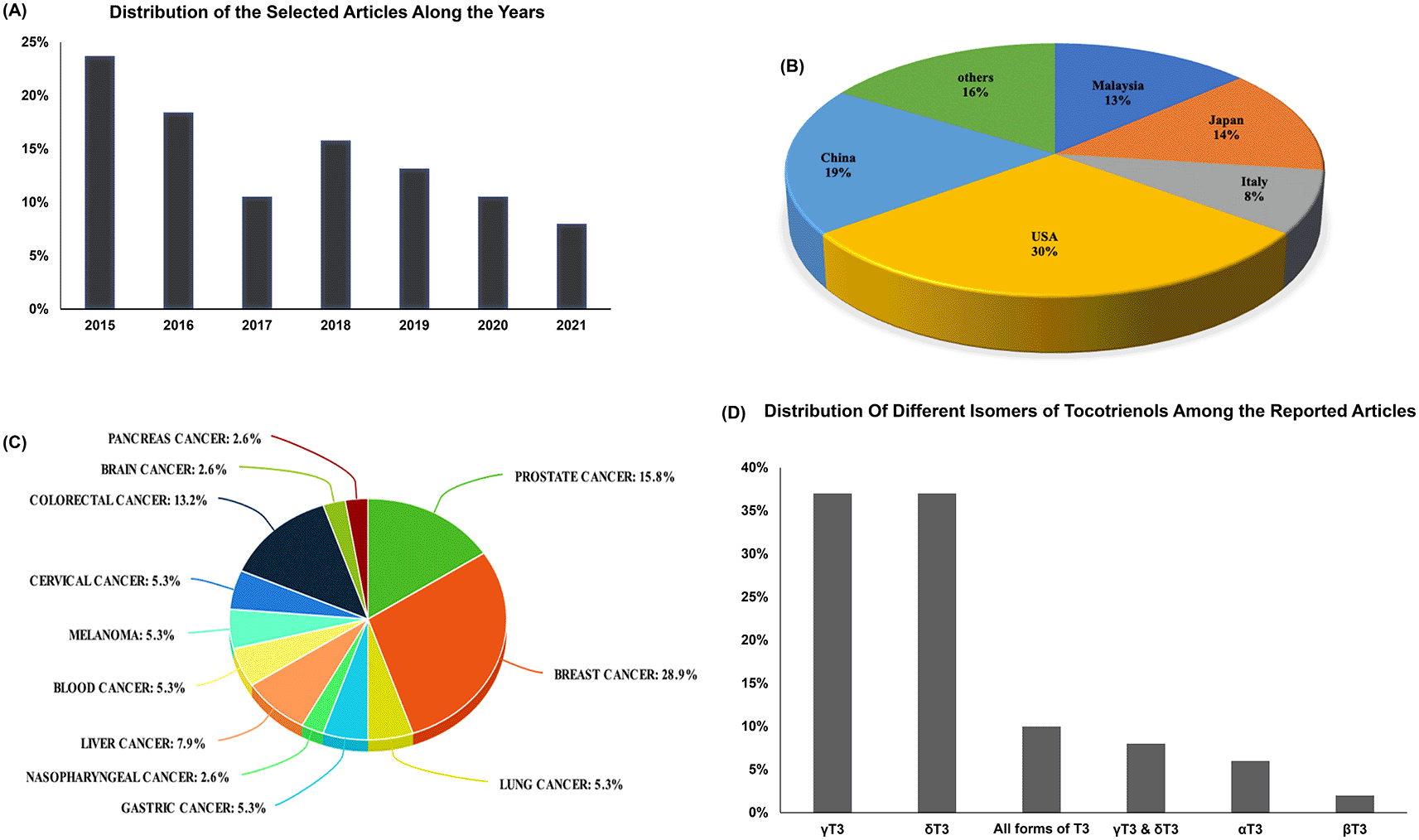
The 37 studies selected for further analysis in this scoping review used several sources of T3. Table 3 shows the source of T3 that most used in the selected studies. More than half of the research papers were published in the years 2015, 2016 and 2017 (53%) (Figure 2A). Interestingly half of the reported studies were conducted in China and the United States of America (Figure 2B)
| Source of Tocotrienols | Tocotrienols | Tocopherols | Reference |
|---|---|---|---|
| Palm oil tocotrienol rich fraction | 70% (all T3 isoforms) | 30% | 6 |
| Rice bran TRF | 61.7% (all T3 isoforms) | 13.1% | 7, 8 |
| Annatto bean oil TRF | 90% δT3 10% γT3 | - | 9 |
| Tocotrienol-rich capsules | 78.7% (all T3 isoforms) | 21.3% | 10 |
| Tocotrienol-rich mixture | 25.8% (all T3 isoforms) | 6% | 11 |
Twelve types of human cancer cell lines were used in the 37 short-listed studies and these include breast cancer,14,26,33–40 prostate cancer,9,11,41–44 lung cancer,10,45 nasopharyngeal cancer,46 hepatocellular carcinoma (liver cancer),47–49 blood cancer,50,51 melanoma], cervical cancer,33,40,54 colorectal cancer,55–60 gastric cancer,61,62 brain cancer57 and pancreatic cancer63 (Figure 2C). The top three human cancer cell lines studied appear to be breast, pancreatic cancer, and colorectal cancers (Figure 2C), while nasopharyngeal and brain cancers were the least studied cancer.
The different isoforms of T3 used in these studies showed prominent anticancer activities (Figure 2D). While most studies used gamma-T3 (γT3) and/or delta-T3 (δT3) from different sources, only one study investigated the effect of beta-T3 (βT3) (Figure 2D). That could be due to the relatively stronger anticancer potential of γT3 and δT3 and/or because of the very low concentration of βT3 in nature.
In general, some of the molecular targets and pathways affected by T3 in humans cancer cell lines appear to be common regardless of the type of the malignancy, such as induction of cell death through apoptosis,9,10,26,33,38,40,42,44–46,49–52,54–57,61,63–65 followed by inhibiting proliferation through inhibition of cell cycle9–11,26,34,35,37,40,43,45–49,54–57,60,61,65 or inhibition of angiogenesis,43,60 cell migration and invasion10,45,60,62,65 (Figure 3). In a smaller number of studies, the T3 appear to exert the anticancer effects through antiglycolytic effect,36 suppressing cancer stem cells,33 antioxidant and synergism with other compounds and antineoplastic therapeutic drugs.50 However, it should be noted that some studies reported more than one mechanim of the cell death mechanisms.

A large number of genes and proteins were differentially regulated in the human cancer cells exposed to various forms of T3. We identified 43 genes/proteins that were reported in in two or more publications (Table 3). Of these 43 genes/proteins, 20 were upregulated and 16 were downregulated. The expresseion of seven of these genes/proteins were contradictory as (Table 3). Gamma and delta-T3 appear to regulate the expression of most of these genes/proteins (Figure 4).
| Gene/Protein | Regulation | Description | References |
|---|---|---|---|
| ALDH2 | ↑ | Aldehyde dehydrogenase, mitochondrial; Aldehyde dehydrogenase 2 family member | 39, 46 |
| AMPK | ↑ | 5'-AMP-activated protein kinase subunit beta-1; non-catalytic subunit of AMP-activated protein kinase | 36, 42, 53 |
| AMPK | ↑ | 5'-AMP-activated protein kinase subunit beta-1; non-catalytic subunit of AMP-activated protein kinase | 36, 42, 53 |
| ATF4 | ↑ | Cyclic AMP-dependent transcription factor ATF-4 | 37, 52 |
| BAX | ↑ | BCL2-Associated X Protein | 37, 44, 46, 52, 54, 63 |
| BIP | ↑ | Binding immunoglobulin protein | 37, 52 |
| CASP3 | ↑ | cysteine-aspartic acid protease (caspase)3 | 14, 37, 44, 46, 52, 54, 55, 57 |
| CASP8 | ↑ | caspase 8 | 40, 51, 57 |
| CASP9 | ↑ | caspase 9 | 40, 44, 51, 54, 55 |
| CDKN1A | ↑ | cyclin dependent kinase inhibitor 1A | 40, 46, 55 |
| CDKN1B | ↑ | cyclin dependent kinase inhibitor 1B | 34, 55 |
| CHOP | ↑ | CCAAT-enhancer-binding protein homologous protein | 37, 52 |
| Cytochrome c | ↑ | cytochrome C | 14, 54 |
| FAS | ↑ | Fas Cell Surface Death Receptor | 40, 51 |
| GSN | ↑ | Gelsolin (protein coding gene) | 39, 46 |
| IRE1Α | ↑ | Inositol-requiring enzyme 1 alpha | 37, 52 |
| JNK | ↑ | Jun N-terminal kinase, also known as (MAPK8) | 37, 42, 53 |
| P38 | ↑ | mitogen-activated protein kinases | 37, 42, 53 |
| P53 | ↑ | tumour protein P53 or TP53 | 46, 51 |
| RAC1 | ↑ | Ras-related C3 botulinum toxin substrate 1 | 39, 46 |
| BCL2 | ↓ | B-cell lymphoma 2 | 14, 37, 40, 46, 51, 52 |
| CDK4 | ↓ | cyclin-Dependent Kinase 4 | 37, 46, 55 |
| c-MYC | ↓ | cytoplasmic -myelocytomatosis oncogene product. | 34, 36 |
| GSK3 β | ↓ | Glycogen synthase kinase 3beta | 34, 40 |
| HES1 | ↓ | hairy and enhancer of split-1 | 10, 45 |
| HIF1A | ↓ | Hypoxia-Inducible Factor 1A | 41, 55 |
| H-RAS | ↓ | Harvey Rat sarcoma virus | 33, 50 |
| MMP9 | ↓ | Matrix Metalloproteinase-9 | 45, 59, 60, 62 |
| MTOR | ↓ | mammalian target of rapamycin | 34, 36, 42 |
| NF-κB | ↓ | Nuclear factor kappa B | 59, 60, 61 |
| NOTCH1 | ↓ | Neurogenic locus notch homolog protein 1 | 10, 45 |
| PI3K | ↓ | Phosphoinositide-3-kinase | 14, 34 |
| SET | ↓ | SET Nuclear Proto-Oncogene | 39, 46 |
| SRC | ↓ | Proto-oncogene tyrosine-protein kinase Src | 9, 48 |
| SURVIVIN | ↓ | baculoviral inhibitor of apoptosis repeat-containing 5 or BIRC5 | 10, 49, 60 |
| VEGFR2 | ↓ | vascular endothelial growth factor receptor-2 | 59, 60 |
| AKT | ↓ | Three closely related serine/threonine- protein kinases (AKT1, AKT2 and AKT3) called the AKT kinase | 34, 36, 50, 55 |
| ↓ | 42 | ||
| ERK | ↑ | extracellular-signal-regulated kinase also known as MAPK1 | 37, 44, 52 |
| ↓ | 33, 50 | ||
| ERK1 | ↑ | member of the mitogen, extracellular-signal-regulated kinase also known as MAPK3 | 34, 37 |
| ↓ | 53 | ||
| GADD45A | ↑ | Growth arrest and DNA damage inducible alpha | 51 |
| ↓ | 46 | ||
| JUN | ↑ | Jun Proto-Oncogene, AP-1 Transcription Factor Subunit (protein-coding gene) | ↑ 44, 63, ↓ |
| ↓ | 40 | ||
| PARP | ↑ | poly-ADP ribose polymerase | 37, 52, 54, 59 |
| ↓ | 10 | ||
| TGFA | ↑ | Transforming Growth Factor Alpha | 35 |
| ↓ | 46 |
The pathway enrichment analysis of these 43 biomarkers was carried out using the Metascape bioinformatic tool66 (https://metascape.org/gp/index.html) (Figure 5). According to the software developers, this software can utilize all genes in the genome for the enrichment background.66 In addition, the software employs several gene ontology sources, including the KEGG pathway, GO Biological Processes, Reactome Gene Sets, PANTHER Pathway CORUM, Wiki Pathways and Canonical Pathways to provide the protein-protein interactions (PPI). To perform the analysis, the 43 biomarkers were uploaded into Metascape. The highest expressed 20 clusters based on enriched terms were used to extract biological information based on their molecular functions and disease bio-pathways curated from KEGG databases, WikiPathways and GO Biological Processes (Figure 5A). Biomarkers with a p-value < 0.01, a minimum count of 3, and an enrichment factor > 1.5 were collected and congregated into clusters according to their membership similarities (Figure 5B and 5C). The kappa scores were used to execute hierarchical clustering on the enriched terms and sub-trees (similarity of > 0.3 are considered a cluster). The cluster name represents the most statistically significant term in the cluster (Figure 5C). The top biological pathways identified in this analysis were cancer pathway followed by apoptosis, gastrin signalling pathway, prostate cancer and VEGFA-VEGFR2 signalling pathway (Figure 5B). The apoptosis pathway contained 23 of the identified genes (PARP1, AKT1, XIAP, BIRC5, FAS, ATF4, BAD, BAX, BCL2, CASP3, CASP8, CASP9, GADD45A, DDIT3, ERN1, HRAS, JUN, PIK3CA, MAPK1, MAPK3, MAPK8, TP53, TUBA1C, CDKN1A, MMP9, MYC, RB1, SRC, GSK3B, RAC1, XBP1, CRK, MTOR, HLA-A, RAC2, CCND1, NFKB2, PRKAA1, LONP1, PSEN2, CDK4, CDKN1B, GSN, DYNC1I2, PTPN6, PTPN11, NOTCH1) while the Gastrin signalling pathway contained 20 genes (AKT1, BIRC5, BAD, CCND1, CASP3, CDKN1A, CDKN1B, CRK, MTOR, GSK3B, HRAS, JUN, MYC, PIK3CA, MAPK1, MAPK3, MAPK8, PTPN11, RAC1, SRC, TP53, BAX, CASP9, ATF4, BCL2, PSEN2, PTPN6, PRKAA1, XIAP, PGR, RB1, RAC2, XBP1, CDK4, GADD45A, DDIT3, HES1, NFKB2, ELF2, PRKAG1, CASP8, MMP9, GSN, ERN1, EPHB2, FAS, PARP1, ATP1A3, TUBA1C, HLA-A, EEF1A1, DYNC1I2, GDF10, NOTCH1) and the VEGFA-VEGFR2 signalling pathway had 24 genes (AKT1, BIRC5, FAS, ATF4, CCND1, BCL2, CRK, EPHB2, ERN1, MTOR, GSK3B, HRAS, JUN, PIK3CA, PRKAA1, MAPK1, MAPK3, MAPK8, PTPN6, PTPN11, RAC1, SET, SRC, TUBA1C). The genes in the VEGFA-VEGFR2 signalling pathway were essential in the regulation of angiogenesis and proliferation of cells.67 Some of the molecular targets that reported to be involved in angiogenesis and cell survival such as AKT1, VGF2, CCND1& HRAS were significantly downregulated by T3 in the human cancer cells.

(αT3: alpha-T3, δT3: delta-T3, γT3: gamma-T3; T3: tocotrienol; TRF: T3-rich fraction) *data from TRF and capsule-rich T3 not included in this Venn diagram.

(A) shows a bar graph of enriched terms across input gene lists, colored by p-values, as shown on the sidebar, while (B) and (C) are protein-protein interaction (PPI) enrichment analysis of the molecular complex detection (MCODE) algorithm has been identified for individual gene lists have been gathered and are shown in (B) the three best-scoring terms by p-value are the functional description of the corresponding components genes are shown in the figure. In (C) MCODE1 represents Chromosomal and microsatellite instability in colorectal cancer and cancer pathways); MCODE2 represents PI3K-AKT-mTOR signaling pathway and therapeutic opportunities and cancer pathways; and MCODE3 represents apoptosis and cancer pathways). The size of the node is related to the protein p-value.
Utilizing the highest ten enrichment analysis pathways that were carried out using KEGG (log P is between -42.7857 and -30.1946) 40 common genes i.e. represented in more than two pathways were selected for protein-protein interaction (PPI) analysis. The 40 proteins were uploaded into an online database that can be used for PPI known as the STRING database ( https://string-db.org). Mapping of the 40 genes showed 40 nodes (interactions) along with 225 edges and 63 expected edges with PPI enrichment p-value:< 1.0e-16 analysed at high confidence (0.700) minimum required interaction score. The selected proteins formed three clusters that contained 21,12 and 7 proteins (Figure 6). The top molecular functional processes with the highest strength were used to determine the functions associated with the annotated proteins. The top functional processes were identified to be histone deacetylase regulator activity (GO:0035033; p= 0.0039, strength: 2.39), cysteine-type endopeptidase activity involved in apoptotic signalling pathway (GO:0097199; p=0.00017; strength: 2.26), BH3 domain binding (GO:0051434; p=0.0064; strength: 2.21), cyclin-dependent protein serine/threonine kinase inhibitor activity (GO:0004861; p=0.00038; strength: 2.09) and MAP kinase activity(GO:0004707; p=0.0011; strength: 1.87). Interestingly, all the molecular functions that regulated initiator cell death were found in one cluster, which includes the mammalian family of mitogen-activated protein kinases (MAPK 1,3 and 8), the caspase family proteins CASP 3,8 and 9, and some cell cycle regulators such as CDK4, CDKN1B and CDKN1A (Figure 6). Based on the STRING analysis, the mammalian family of MAPK appear to be important targets for T3s to exert their anticancer activity, which is in sync with their pro-apoptotic and cell cycle dysregulation activities. However, MAPKs activity is subjective to the type of the malignancies, and their microenvironment, as these have been found to have dual effects as oncogenes and tumour suppressors in some cancers and related microenvironment.68,69

The dotted line indicates the interactions among the proteins in the three clusters.
The potent cytotoxic effect of T3s is explored and evident in several cancers, yet the exact route through which this action is expressed is still not fully elucidated. In this scoping review, we attempted to understand how T3 exerted their anticancer activity in human cancers and the regulatory mechanisms that govern the anticancer activities. The findings from this study strongly suggest that the most common means by which T3 mediate their effect were through induction of apoptosis and inhibition of proliferation, angiogenesis, and metastasis.
In this review, 25 articles reported that T3 promoted anticancer effects through cell cycle arrest and/or dysregulation of pathways that control cell proliferation and death. One such mechanism was through activation of deoxidation due to overexpression of the PRX4 protein, which causes reduction of hydrogen peroxide and alkyl hyperoxide into water and alcohol and activation of NF-kB.47 Another mechanism was the ability of T3 to inhibit the expression of the telomerase reverse transcriptase (TERT) gene in cancer cells. This TERT gene is an essential gene and protein that is involved in cellular proliferation and genetic transcription, which is found in more than 80% of most tumours but not in healthy cells.70 Delta T3 was reported to suppress the expression of the TERT gene in human colorectal cancer cells.56 In addition, the cytotoxic effects of T3 can be mediated by decreasing the expression of the hypoxic inducible factor-1α (HIF-1α) gene in cancer stem cells and in androgen-resistant prostate cancer.41 It should be noted that HIF-1α is highly expressed in many malignancies and this gene is responsible for various regulatory mechanisms, including hypoxia, angiogenesis and cell proliferation.41 The antiproliferative effect of T3 on cancer cells is most likely mediated through suppression of proliferative and cell survival pathways such as the VEGFA-VEGFR2 signalling pathway and RAC1/PAK1/p38/MMP2 pathway and activation of apoptosis.
The cell cycle consists of four phases; S phase (DNA synthesis) and M phase (mitotic phase), which are set apart by two gaps (G) phases, G1 and G2 (Figure 7). The cell cycle is regulated by cyclins, which are regulatory blocks of cyclin dependent kinases (CDK) that form heterodimers that promote proliferation and govern the cell cycle checkpoints. The CDK regulate genomic and chromosomal activity in many neoplasms leading to the uncontrolled proliferation and tumorigenesis of cancer cells. The cell cycle has many checkpoints and various types of cyclins are synthesized and dispersed at the cell cycle checkpoints. Checkpoints are regulated by CDK-cyclin complex inhibitors such as p16, p18, p21 and p27, which control downstream activation of the cell cycle pathways, where over-expression can result in cell cycle arrest.71 The two crucial checkpoints are (i) after DNA replication midway to the end G1 phase before DNA replication and (ii) at G2 before chromosomal segregation. The first checkpoint is regulated by CCND1, which is the most common CDK, that phosphorylates the retinoblastoma protein (pRB), allowing the cell cycle to progress while unphosphorylated RB suppresses cell proliferation and keeps the cells at G0.72 The CCND1 is overexpressed in several human malignancies such as breast cancer, lung cancer, sarcomas and squamous cells.73 The p21(CDKN1A) and p27 (CDKN1B) are key suppressors of CCND1 and crucial to activating cell cycle arrest at G1.72 Dysregulation of CDKs is an essential feature of many cancers and provides good targets for drug discovery. Over-expression of CDK in several cancers have become essential targets for drug therapy, such as CDK4/6 inhibitors ribociclib and abemaciclib.74,75

T3s regulated the molecular targets demonstrated in this diagram in two or more studies.
In prostate cancer cells, T3 suppressed proliferation and induced apoptosis through dysregulation of several molecular targets such as NF-κB, AKT, STAT3, p53 and p21.9,11,72,76 The T3 were reported to increase the expression of p21 and p27 and suppress histone deacetylases in human prostate cancer cells.11 Similarly, the anticancer effect of δT3 on nasopharyngeal cancer (NPC) was mediated through several molecular targets, including downregulation of p21 and upregulation of p16, and downregulation of CCND1, CDK2 and CDK4, which caused cell cycle arrest, possibly due to dysregulation of P16/CDK4/CCND1 signalling pathway.46 In this review, we found that the members of the cyclin protein family were often downregulated by T3s regardless of the malignancy origin.11,34,46,49,60 In addition, exposure to T3 caused upregulation of p21 and p27 in several human cancer cell lines11,34,46,55 except in one study, in which γT3 and δT3 suppressed the expression of the p21 gene in cervical cancer cells.40
The extrinsic pathway of apoptosis is regulated by the death receptors such as the tumour necrosis factor receptors (TNFR), tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) and the TNFR superfamily member-6 (TNFRSF6/FAS). These receptors regulate apoptosis, thus, the fate of the cell. The intrinsic pathway of apoptosis is coordinated by the mitochondria and is set in motion by different proteins that mediate and control cellular homeostasis. One of cancer leading survival mechanisms is decreased expression of pro-apoptotic proteins and increased expression of anti-apoptotic proteins.77 An example of that critical pro-survival proteins is the BCL2 family, which inhibits several pro-apoptotic proteins that exhibit the homology domain of the BCL2 protein, such as BAX and BAK. Overexpression of BCL2 in cancers is associated with treatment resistance and poor prognosis.78 Another important protein is the p53 tumour suppressor gene. Lack of p53 allows the progression and proliferation of damaged genetic material. However, activation of p53 upregulates the expression of several genes, such as p21.79 It can also activate BAX, an essential gene regulator of apoptosis in both intrinsic and extrinsic pathways.80,81 In this review, six research articles reported that exposure to T3 increased the expression of p53 and BAX in human cancer cells (Figure 8). In addition, the T3 isoforms inhibited the expression of BCL2 in various cancers triggering the expression of several pro-apoptotic proteins and genes causing cell death.14,37,40,46,51,52 For example, in cervical cancer cells, T3 reduced the expression of BCL2 and induced apoptosis by upregulating BAX and cytochrome C, a cascade of molecular events expressed during mitochondrial apoptotic pathway activation.54 In NPC cells, γT3 decreased the expression of the BCL2 gene and BCL2 protein, which in turn activated BAX2 and upregulated CASP proteins 3,8 and 9, causing activation of both apoptotic pathways.46 FAS, a membrane protein belonging to the death receptors (DR) family, can induce apoptosis by triggering the caspase cascades (Figure 8). It was reported that exposure to δ-T3 induced cytotoxic cell death in chronic amyloid leukaemia that was mediated by the overexpression of FAS and activation of the extrinsic apoptotic pathway, among other reported genes that are related to the activation of apoptosis.51 The pro-apoptotic activity of T3 can be mediated through action on different molecular targets that regulate apoptotic cell death (Figure 8).
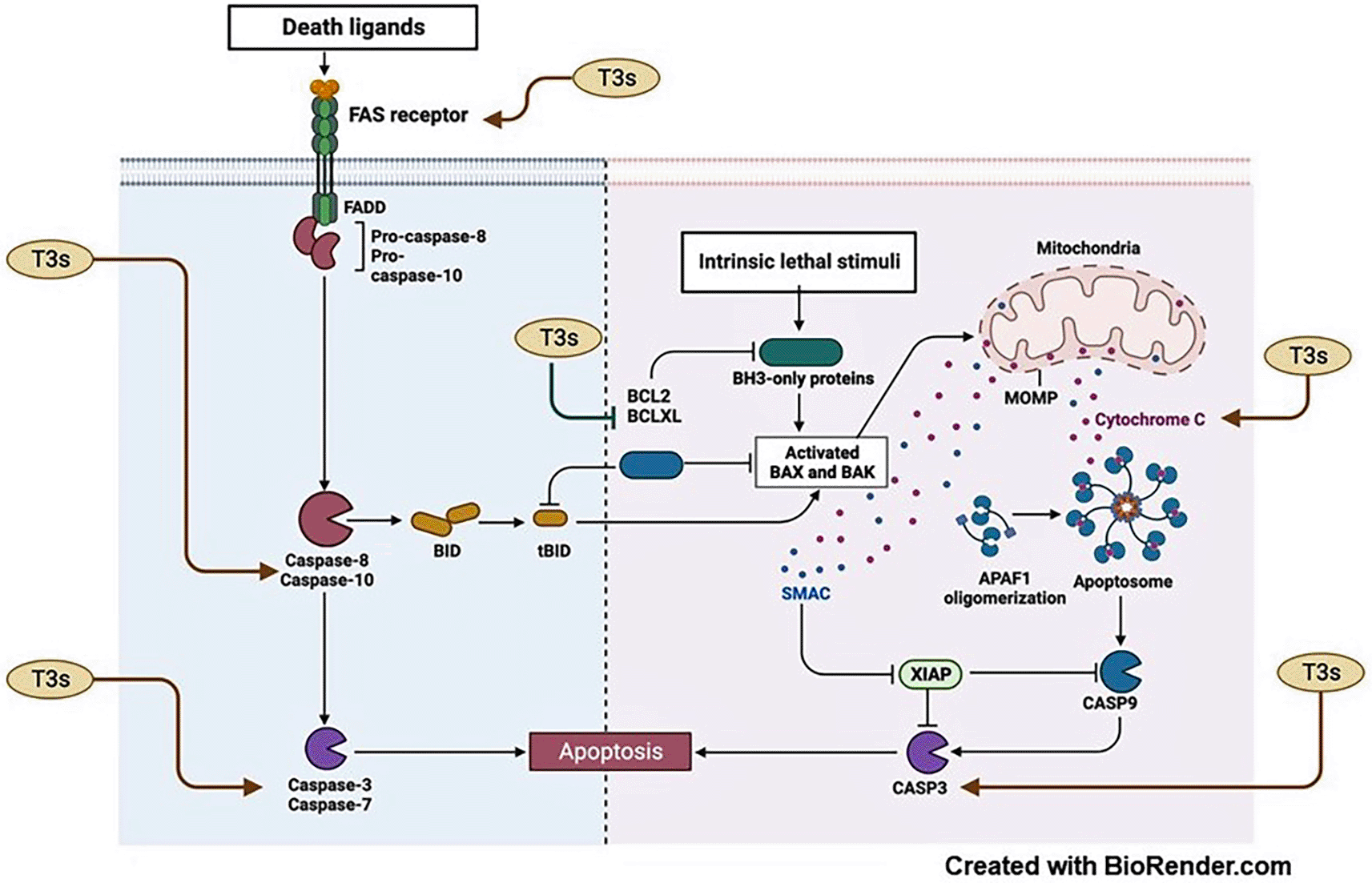
One of the primary mechanisms through which solid tumours survive is their ability to form new vasculature and, thus, gain the ability to proliferate at a higher rate and migrate to a suitable microenvironment. Forming new vessels (angiogenesis) is fundamental for tumour proliferation, sustained nourishment, and oxygen supply. Hypoxia-inducible factor (HIF) is a major factor in the activation of angiogenesis and cancerous cell migration, in search for a better microenvironment. Tumours express abundant amounts of HIF, which in turn regulates the vascular endothelial growth factor (VEGF) and the VEGF receptor transcription responsible for new vasculature.82 Several anticancer therapies target the molecular signalling pathways responsible of the formation of new vessels to decrease oxygen and nutrition supply to tumours, which will eventually lead to the death signal’s activation and subsequently killing the cancerous cells. Many known treatments exert their cytotoxic action via these processes, such as Sunitinib inhibitor of VEGF receptor tyrosine kinase activity and Bevacizumab, a selective inhibitor of circulating VEGF used in the treatment of refractory ovarian cancer.83 In this review, the antiangiogenics activity of T3 was among the five most common cellular mechanisms through which T3 exert their actions. Similarly, key molecular targets in tumour angiogenesis, such as HIF1A and VEGFR2, were superseded by T3.
A limitation of this study is that it targeted all types of human cancer cell lines to find common molecular targets and did not target on a single type of human cancer. In addition, the review may have missed papers that did not meet the time scope of this review.
As reported by the reviewed studies, T3 regulate several underlying cancer cell death mechanisms in different cancer cell lines. Such as a cell cycle regulator and apoptosis inducers. Further targeted studies are required to map the definite pathways by which T3 exerts their action. The reported potent antineoplastic effects in cell culture-based studies and animal studies invite extensive examination of the common mechanism by which T3 exert their action in order for this promising natural compound clinical use.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)