Keywords
Carcinoids, whole exome sequencing, Whole transcriptome, Splicing variants, Small bowel carcinoids, Metastatic carcinoids, Small intestine neuroendocrine tumors, Metastatic small intestine neuroendocrine tumors, SI-NETs, Metastatic SI-NETs
This article is included in the Genomics and Genetics gateway.
Carcinoids, whole exome sequencing, Whole transcriptome, Splicing variants, Small bowel carcinoids, Metastatic carcinoids, Small intestine neuroendocrine tumors, Metastatic small intestine neuroendocrine tumors, SI-NETs, Metastatic SI-NETs
Overall: We have updated our text to more frequently use the latest terminology “SI-NETs.” We also added this terminology to the manuscript’s “keywords” list. We have cited 11 additional papers (several of which were published after we had written our initial text). We italicized the names of a handful of genes and bioinformatic tools (where the formatting did not transfer correctly in ver1).
Abstract: We changed some phrasing to improve flow and terminology use.
Introduction: We added additional citations; improved flow and terminology.
Methods: We added an explicitly-stated "limitations" section, per one reviewer's request. We specified the origin of the constitutional/normal tissue samples. We highlighted the fact that RNA data was not available for normal tissue at the time of the study. Per a reviewer’s request, we mentioned that the iDEP workflow employs parametric and semi-parametric tools.
Results: We specified the treatment sequences (i.e. surgery followed by chemotherapy) for patients more clearly (also in adjoining Table 1). We cited additional relevant papers.
Discussion: We used the terminology "SI-NETs" more frequently.
Figures & Tables: We corrected Table 1. We italicized the genes in the top row of Table 3 (formatting had not transferred correctly in ver1). We reworded the caption of Figure 5, and removed Table 5.
Citations: We added 11 citations.
Keywords: We added 4 keywords re: "SI-NETs."
See the authors' detailed response to the review by Phillip Stafford
See the authors' detailed response to the review by Anela Blažević
Small intestine neuroendocrine tumors (SI-NETs) are the most common malignancy of the small bowel (Bilimoria et al. 2009). These tumors are characterized by their secretion of biogenic amines (such as serotonin and histamine), peptides (such as somatostatin and chromogranins), tachykinins, and/or prostaglandins (Pinchot et al. 2008; de Mestier et al. 2021). Secretion of these substances can lead to carcinoid syndrome, which is characterized by diarrhea, abdominal pain, bronchoconstriction, skin flushing, and valvular heart disease (Eltawil et al. 2022; Tran, Sherman, and Howe 2021; Moertel 1987). For this reason, SI-NETs have historically also been referred to as “carcinoid” tumors.
It has been postulated that SI-NETs are derived from enterochromaffin cells within intestinal crypts (Pinchot et al. 2008). Anatomically, small bowel carcinoids most frequently occur in the terminal ileum (Keck et al. 2018). Although these tumors tend to be indolent and insidious, they are often already metastatic at the time of diagnosis (Yao et al. 2008; Shah et al. 2019). The median age of diagnosis for small bowel carcinoid tumors is 61 years (Shah et al. 2019). Carcinoids are generally well-differentiated and slow-growing (Cunningham et al. 2011; ‘StatPearls’ 2022). These tumors most often spread to regional lymph nodes, adjacent mesentery, and the liver (Pinchot et al. 2008). Improved imaging modalities, including the intravenous use of radiopharmaceutical gallium 68Ga-or copper 64Cu-dotatate paired with positron emission tomography (PET), may demonstrate metastases that are not appreciated with other, less sensitive imaging studies (Sanli et al. 2018).
Surgical removal of a localized, primary SI-NET (along with adjacent mesentery and lymph nodes) can be curative (Quinones et al. 2022). Treatment of metastatic carcinoid tumors, however, requires selection from a repertoire of therapeutic options, including surgical resection, administration of somatostatin analogues, peptide receptor radiotherapy with 177Lu-dotatate, hepatic arterial embolization, hepatic radiofrequency ablation, external beam radiotherapy of selected isolated metastases, and administration of targeted therapies (Howe et al. 2017).
As the incidence of small bowel carcinoid tumors is increasing at an annual rate of 6.3%, the development and adoption of more effective targeted therapies is imperative (Ahmed 2020; Dasari et al. 2017; Maggard, O'Connell, and Ko 2004). Better elucidation of molecular drivers of carcinogenesis and metastasis underlying SI-NETs will help foster the development of new treatments. Knowledge of the full genomic landscape of small bowel carcinoid tumors is limited, as prior studies have deemed them to be relatively “mutationally silent” compared to other malignancies (Elias et al. 2021; Lim and Pommier 2021; Priestley et al. 2019). Loss of chromosome 18 as well as various loss of function mutations in cyclin-dependent kinase inhibitor 1B (CDKN1B, which encodes the cell cycle regulatory protein p27) have been reported in a minority of patients (i.e. 9%) (Banck et al. 2013; Crona et al. 2015; Crona and Skogseid 2016; Cunningham et al. 2011; Francis et al. 2013; Kulke et al. 2008; Kytölä et al. 2001; Lim and Pommier 2021; Löllgen et al. 2001; Samsom et al. 2021; Zhang et al. 2020).
Reported aberrations include germline mutations in CDKN1B, which are known to cause Multiple Endocrine Neoplasia type IV (MEN4) (Seabrook et al. 2022). It has been suggested that loss of chromosome 18q may be an early event in the evolution of SI-NETs, whereas loss of CDKN1B and, therein, loss of tumor suppressor p27, occur later in malignant progression (Crona and Skogseid 2016; Di Domenico et al. 2017).
Recent studies have demonstrated that analysis of RNA may reveal the presence of causative molecular drivers of disease when DNA-level analysis has failed to do so (McCullough et al. 2020). Therefore, the integration of genomic data with transcriptomic data is the next logical step toward characterizing the molecular alterations underlying SI-NETs and, in doing so, identifying biomarkers for precision diagnosis, prognosis, and treatment. In this study, we investigated the genomic and transcriptomic landscapes of trio sets of germline, primary tumor, and metastatic tumor samples derived from 5 patients. We observed consistent loss of chromosome 18, as well as loss of function (LOF) mutations in CDKN1B. We identified metastasis-specific mutations in primary and metastatic carcinoid tumor pairs, several of which have been reported as driver mutations in other neuroendocrine tumor types. Transcriptome sequencing added relevant information that would not have been appreciated from DNA data alone. The detection of several splicing mutations on the DNA level, and of their consequences at the RNA level, suggest that RNA splicing aberrations may be an important mechanism underlying the development of SI-NETs.
Five patients (here referred to as 0006, 0007, 0008, 0009, 0018) undergoing surgical resection of their carcinoid tumors at Hoag Hospital (Newport Beach CA) or the University of Iowa Neuroendocrine Tumor Clinic consented to this study (written consent), which received IRB approval from the participating institutions (Hoag IRB number: 20180303; Iowa IRB number: 199911057). These patients had samples available from normal/constitutional tissue (i.e., gallbladder for patients 0006 and 0007, healthy bowel for patients 0008 and 0009, and peripheral blood for patient 0018), primary SI-NETs, and liver metastases from time of surgical resection. Deidentified tissue samples were sent for DNA/RNA extraction and sequencing as detailed below at the Translational Genomics Research Institute (TGen) (Phoenix AZ).
Tumor or constitutional DNA was extracted using the Qiagen AllPrep Kit or GeneRead FFPE DNA Kit (Cat. No. 80234, 180134), and tumor RNA was extracted using the Qiagen AllPrep Kit or RNeasy Mini Kit (Cat. No. 74104). Using 200 ng of input DNA, whole exomes were constructed for each sample using the Kapa Hyper Prep Kit (Cat. No. 07962363001) using Agilent SureSelect Human All Exon V7 baits. RNA libraries were constructed using 500ng of input RNA per sample and using the Illumina TruSeq Stranded Total RNA Kit with Ribo-zero (Cat. No. RS-122-2201). Paired-end sequencing of libraries was performed on the Illumina NovaSeq 6000 using S1 and SP flowcells for 100bp reads. Approximate sequencing targets were 200× for tumor exomes, 100× for constitutional exomes, and 150 million total reads for each RNA library.
Paired DNA and RNA data (for germline, primary, and metastatic tissue samples) were input into two pipelines: TGen’s Phoenix pipeline, and the Keck School of Medicine of USC’s Genomics Platform (KGP) “Echo” pipeline.
This pipeline utilizes SAMtools (v1.10) and Burrows-Wheeler Aligner (bwa v0.7.17) for alignment of whole exome sequencing (WES) data; STAR (v2.7.5a) for RNA sequencing (RNAseq) alignment; DeepVariant (0.10.0-gpu) to call germline SNVs and small indels; lancet (v1.1.x) for somatic variant calling; Manta (v1.6) for detection of somatic structural variants and indels; Octopus (v0.6.3-beta) for haplotype-based variant calling; Strelka2 (v2.9.10) to call small somatic variants; Vardict (Java v1.7.0), which calls somatic SNVs, multi-nucleotide variants, indels, structural variants, and loss of heterozygosity; SnpEff (v4.3T) for SNP annotations; and vcfMerger2 (v0.8.7).
Whole exome FASTQs (tumor/normal pairs, for both primary and metastatic tumors) were aligned to human genome build GRCh38 (Gencode v29 primary assembly) using bwa mem (v0.7.17). Base quality score recalibration was performed using GATK’s BaseRecalibrator (v4.0.10.1) and ApplyBQSR. SAM files were merged, and duplicate reads were marked using GATK’s MergeSamFiles and MarkDuplicates, respectively. Quality control metrics for the resulting sorted, indexed BAMs were retrieved using Samtools Stats, Picard HSMetrics, Picard GCBias Metrics, and Picard MultiMetrics. Variants and mutations were called using a dbSNP reference variant call format (VCF) file (v146 hg38) and GATK HaplotypeCaller. Resulting VCFs were annotated with GATK’s Mutect2 (for detection of somatic point mutations), Manta (v1.5.0, for detection of structural variants and indels), Strelka (v2.9.0, for detection of somatic SNVs and small indels), TGen’s Seurat (v2.5, for detection of somatic point mutations), TGen’s tCoNuT (v1.0, for copy number analysis), TGen’s translocation tool, Freebayes (v1.2.0, a Bayesian small SNV/indel detector), and SnpEff (predictor of variant effects). The final variant candidate set was annotated with Ensembl Variant Effect Predictor (VEP).
RNAseq FASTQs (tumors only) were aligned to human genome build GRCh38 using STAR (v2.6.1d). Duplicates were marked using GATK’s MarkDuplicates. Gene fusion predictions were made using STAR-Fusion (v2.6.1d). Quality control metrics for the resulting sorted, indexed BAMs were retrieved using Samtools Stats, Picard RNA Metrics, and Picard MultiMetrics. Variants and mutations were called using a dbSNP reference VCF (v146 hg38) and GATK’s RNA HaplotypeCaller. Salmon, featureCounts, HTSeq Counts, and Cufflinks were used for gene quantification. The final variant candidate set was annotated with VEP. We utilized MultiQC to compile analysis logs and create a comprehensive quality control report. Differential expression and pathway analyses were run on primary/metastatic tumor pairs (not on germline, as constitutional transcriptomic data was not available) using iDEP (which employs the parametric and semi-parametric packages limma and DESeq2) with GAGE analysis, KEGG gene sets, and Pathview for visualization (Ge, Son, and Yao 2018; Luo and Brouwer 2013; Kanehisa et al. 2021).
The authors acknowledge the small sample size of this study, which is not a meta-analysis, and suggest that readers interpret results (particularly those of differential expression and pathway analyses) within the context of this limitation. Sample size appears to be a common limiting factor for genomic studies on SI-NETs (Wu et al. 2022). Many of these previous studies did not utilize RNA data, however, so one of the advantages of this present study is that it is among the first to leverage the transcriptome for identification of molecular drivers.
Tumor samples had, on average, a 45-fold gene enrichment, a coverage ≥30× for 95% of target bases, 87% aligned reads, and 96% RNAseq correct strand mapping. Constitutional samples had, on average, a 43-fold gene enrichment, a coverage of ≥30× for 91% of target bases, and 99.8% aligned reads.
Patient 0006 is a 66-year-old male with grade 1 disease, treated with surgical resection then Octreotide (Table 1). He had large-scale copy number loss of chromosome 18 (Table 2). He also had a PYGL mutation (rs74464749) in both his primary and metastatic tumors (Table 3). He had a metastasis-specific mutation in MXRA5 (chrX:3,317,443:G:C).
Patient 0007 was a 62-year-old female with grade 2 disease (for both primary and metastatic tumors), treated with surgical resection then 177Lu-dotatate (Table 1). She succumbed to her disease. She had large-scale copy number changes in both her primary and metastatic tumors; these included: gain of chromosomes 5, 7, 10, 14q, 15q, and 20, and loss of chromosomes 18 and 11q (Table 2). Loss of chromosome 11q has previously been noted as a potentially metastasis-related event in primary/metastatic SI-NET pairs (Alexander and Ziv 2023; Elias et al. 2021). Amplification of chromosomes 7, 14, and 20 has also been reported in ileal neuroendocrine tumors (Mäkinen et al. 2022). This patient did not have any apparent pathogenic point mutations.
Patient 0008 was a 67-year-old male with grade 2 disease, treated with surgical resection, everolimus, then temozolomide (Table 1). He succumbed to his disease. He had one copy number alteration observed in both his primary and metastatic tumors, gain of chromosome 5 (Table 2). He also had metastasis-specific copy number changes including gain of chromosomes 7 and 10, and loss of chromosomes 9 and 18. As mentioned for patient 0007, amplification of chromosome 7 has been observed in ileal carcinoids (Mäkinen et al. 2022). Loss of the p-arm of chromosome 9 has been previously reported in midgut neuroendocrine tumors (Alexander and Ziv 2023). He had metastasis-specific CDKN1B (rs797044482) and a UBE4B (chr1:10,105,515:G:A) mutations (Table 3).
Patient 0009 is a 52-year-old female with grade 1 disease (for both primary and metastatic tumors, grade 2 lymph nodes), treated with surgical resection (Table 1). The only copy number alteration observed in her tumors was loss of chromosome 18 in the primary/metastatic pair (Table 2). An ATRX point mutation (chrX:77,683,393:G:T) was identified in both her primary and metastatic tumors resulting in loss of heterozygosity (LOH) and allele-specific expression (ASE) in the mRNA (monoallelic expression of the wild-type allele in mRNA) (Table 3). She had a metastasis-specific mutation in SMARCA2 (rs752254994).
Patient 0018 is a 71-year-old female with grade 2 disease, treated with surgical resection then monthly lanreotide (Table 1). She had copy number loss of chromosome 18 in both her primary and metastatic tumors (Table 2). She had metastasis-specific, large-scale copy number loss of chromosome 16q, which has previously been reported in the literature as a metastasis-specific event (Alexander and Ziv 2023). She also had a metastasis-specific ATRX mutation (chrX:77,684,450:T:A) with monoallelic expression of the wild-type allele in mRNA (Table 3).
The results of our analysis performed with iDEP’s GAGE tool and KEGG gene sets suggested that the top 20 pathways with differentially expressed genes between metastatic vs. primary SI-NETs included: “pathways in cancer,” “chemical carcinogenesis,” and “viral carcinogenesis” (Figure 5; Table 4). The pathways within the “pathways in cancer” framework that appear to have the most significant changes in expression between metastases vs. primary tumors include: cytokine-cytokine receptor interaction, p53 signaling, extracellular membrane receptor and focal adhesion interactions, Wnt signaling, PI3K-Akt signaling, MAPK signaling, calcium signaling, TGF-b signaling, HIF-1 signaling, Notch and Hedgehog signaling, estrogen and androgen signaling, cell cycle, and block of differentiation (Figure 5). A recent study also demonstrated that Wnt signaling and focal adhesion pathways were associated with SI-NETs (Chen et al. 2023); however, this same study highlighted cytokinesis, iron ion homeostasis, platelet degranulation, and several metabolic processes to be differentially upregulated in SI-NET metastases compared to primary tumors (which we did not observe).
Previous genomic analyses have failed to identify a consistent, putative driver mutation in small bowel neuroendocrine tumors. We endeavored to dive deeper into the molecular landscape of SI-NETs by supplementing exome analysis with transcriptomic analysis in paired primary small bowel tumors and liver metastases from the same patients. We were particularly interested in discovering drivers of metastatic potential, as metastatic SI-NETs pose the greatest morbidity and mortality risks. We intended to probe RNA for potential culprits that were, perhaps, not immediately evident from DNA analysis alone. RNAseq data enabled us to confirm the consequences of alterations found on the DNA level while also observing phenomena (like intron retention and ASE) that would not otherwise be apparent.
Tumor cells gain oncogenic and lose tumor-suppressive functions via various mechanisms, including point mutations, CNAs, and structural variants. In this study, we observed various CNAs, the most frequent being loss of chromosome 18. Loss of 18q has been previously reported as a potential mechanism of early oncogenesis in SI-NETs (Crona and Skogseid 2016). The long arm of chromosome 18 spans several confirmed and putative tumor suppressors, including Retinoblastoma-Binding Protein 8 (RBBP8), SMAD family members 2 and 4 (SMAD2/4), and deleted in colorectal cancer (DCC) (Cunningham et al. 2011; Lim and Pommier 2021). Though several studies have suggested that DCC may be the key tumor suppressor lost, this has not been rigorously confirmed (Löllgen et al. 2001; Nieser et al. 2017). Other CNAs – which were less consistent across our samples, but which reflect previously reported evidence – included loss of chromosomes 9p, 11q, and 16q, and gain of chromosome 14 (Table 2) (Cunningham et al. 2011).
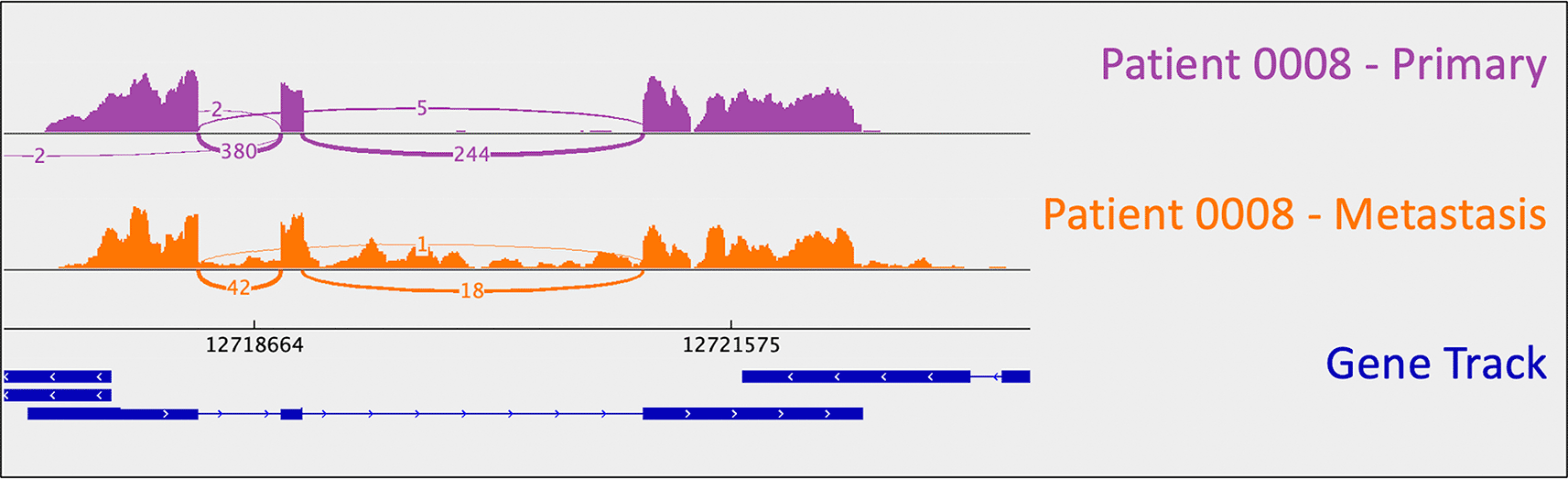
Loss of CDKN1B function is another previously reported characteristic of small bowel carcinoid tumors (though it has only ever been reported in a minority, i.e. 9% of tumors in larger studies) (Crona and Skogseid 2016; Crona et al. 2015; Maxwell et al. 2015). An insertion in CDKN1B (rs797044482, reported as “likely pathogenic” for neuroendocrine neoplasms in ClinVar) was identified in patient 0008’s metastasis (Figure 1). Indel presence was confirmed in mRNA. Interestingly, this patient had a “second hit” to CDKN1B: uniparental disomy (and resulting LOH). B-allele frequency (BAF) is a measure of allelic balance, whereby a heterozygous SNP would have a BAF of 0.5 and a homozygous SNP – or LOH event – would result in a BAF that deviates from 0.5 (to 0 or 1). Patient 0008’s uniparental disomy event was identified via CNA, as B-allele frequency deviated from 0.5 despite the fact that there was no copy number change at that location. This example exemplifies the ability of cancer cells to “mix and match” mechanisms to gain and lose gene products.
A loss-of-function frameshift mutation resulting in intron inclusion is observed in the metastatic tumor alone. This is perceptible as the orange signal that reads through intronic regions (i.e., the thin blue lines in the gene track). Sashimi plots were generated using the Broad Institute’s Integrative Genomics Viewer (IGV).
The tumor suppressor gene ATRX encodes a chromatin-remodeler that is a member of the SWI-SNF family of proteins; it is involved in transcriptional regulation, DNA recombination, nucleosome remodeling, and DNA repair (Bradley et al. 2019; Valenzuela et al. 2021). Somatic mutations in this gene have frequently been reported in gliomas, gastro/pancreatic neuroendocrine tumors, pheochromocytomas, and paragangliomas (Crona and Skogseid 2016; Jiao et al. 2011). ATRX has also been found to be mutated in gliomas and acute lymphoblastic leukemia (ALL) (Bradley et al. 2019). ATRX plays an essential role in brain development and is ubiquitously expressed at high levels in brain tissue (Valenzuela et al. 2021).
Though conflicting evidence exists, it has been reported that ATRX escapes X-inactivation, perhaps in a developmental stage and/or tissue-specific manner, with ATRX showing biallelic expression in XX-females and monoallelic expression in XY-males (Valenzuela et al. 2021). ATRX deficiency results in impaired nonhomologous end-joining and genomic instability (Bradley et al. 2019). Notably, ATRX mutations are frequently observed in female gastric cancer patients with high microsatellite instability (MSI), tumor mutational burden (TMB), and programmed death-ligand 1 (PD-L1) expression; these characteristics are purported to be predictive biomarkers for immunotherapy response (Ge et al. 2021).
Two of our patients (0009, 0018) had ATRX mutations. Patient 0009, an XX-female, had LOF mutations in ATRX in both her primary and metastatic tumors. Patient 0018, another XX-female, harbored a metastasis-specific ATRX mutation. Curiously, both mutations were predicted to be “tolerated” by VEP and showed ASE/monoallelic expression of the wild-type allele in RNA.
As mentioned above, one metastasis-specific finding was patient 0008’s CDKN1B mutation (rs797044482) (Figure 1). A metastasis-specific missense mutation in the gene Matrix Remodeling Associated 5 (MXRA5, chrX:3,317,443:G:C) was seen in patient 0006 (Figure 2). MXRA5 is a purported tumor suppressor gene (Tegally et al. 2020; Xiong et al. 2012; Cheng et al. 2017). It also plays a role in normal matrix remodeling and anti-inflammatory responses, the disruption of which is essential to metastatic progression (Poveda et al. 2017). This deleterious MXRA5 mutation resulted in the activation of a cryptic splice site and loss of mRNA transcripts. As patient 0006 is male, there was no dosage compensation for MXRA5. Patient 0006’s primary and metastatic tumors had a splicing mutation in liver glycogen phosphorylase (PYGL, rs74464749), resulting in intron retention and expression of alternative splice variants in mRNA. A metastasis-specific missense mutation in SWI/SNF Related, Matrix Associated, Actin Dependent Regulator of Chromatin, Subfamily A, Member 2 (SMARCA2, rs752254994) was called for patient 0009 (confirmed in mRNA, Figure 3). SMARCA2 is part of a chromatin-remodeling complex, and mutations in this gene have been reported in neuroendocrine tumors of the lung and thymus (Fernandez-Cuesta et al. 2014). A metastasis-specific mutation in Ubiquitination Factor E4B (UBE4B, chr1:10105515:G:A) was called for patient 0008, which resulted in intron retention in the mRNA (Figure 4); this was paired with a “second hit” of large-scale copy loss of chromosome 1. Mutations in UBE4B have previously been associated with neuroblastoma, another type of neuroendocrine tumor (Byron et al. 2016).
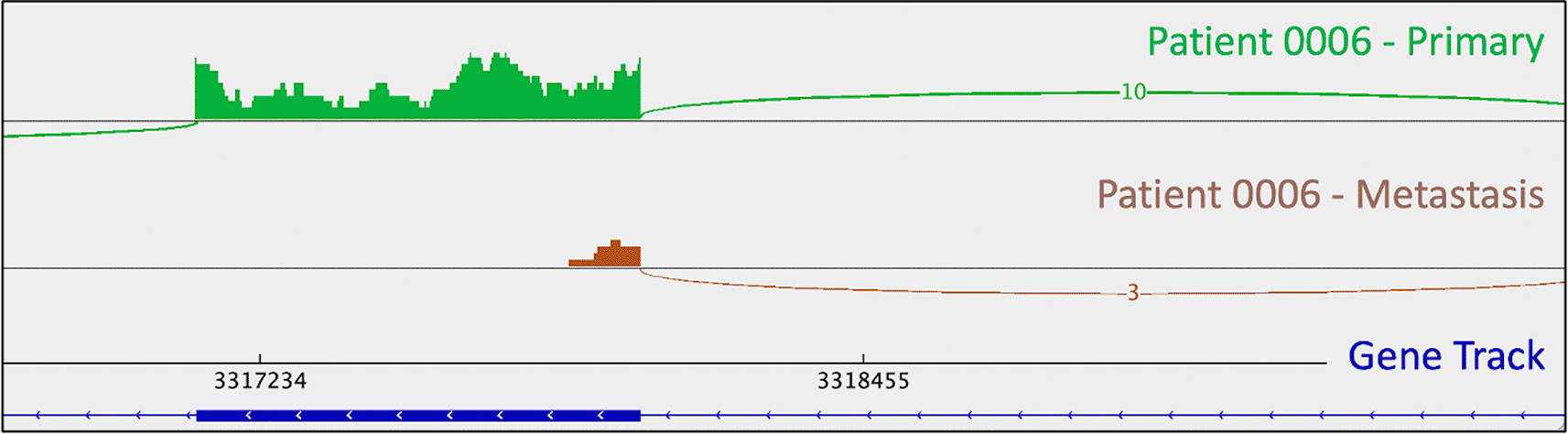
This is appreciable as a lack of mRNA reads corresponding to the downstream portion of the depicted exon (i.e., the thick blue line in the gene track) in the metastatic tumor (brown trace). This phenomenon is not observed in the primary tumor. Sashimi plots were generated in IGV.
Sashimi plots were generated in IGV.
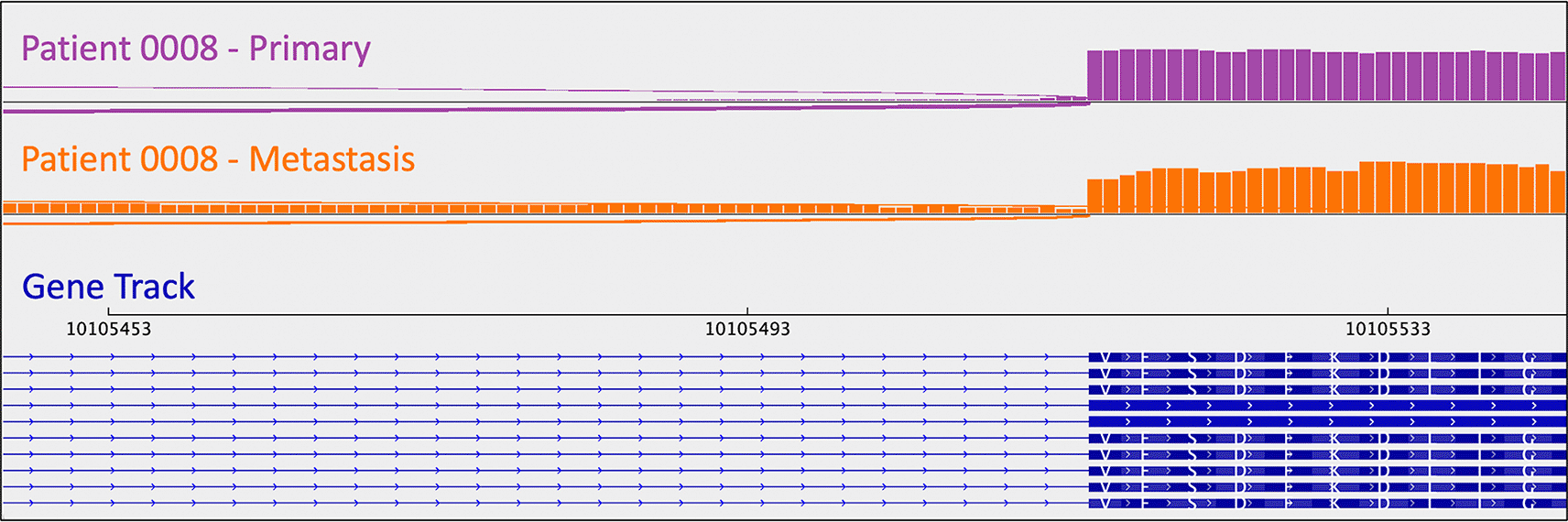
Intron retention is apparent as the presence of mRNA (i.e., the orange signal/bars) corresponding to intronic material (i.e., the thin blue lines in the gene track). Sashimi plots were generated in IGV.
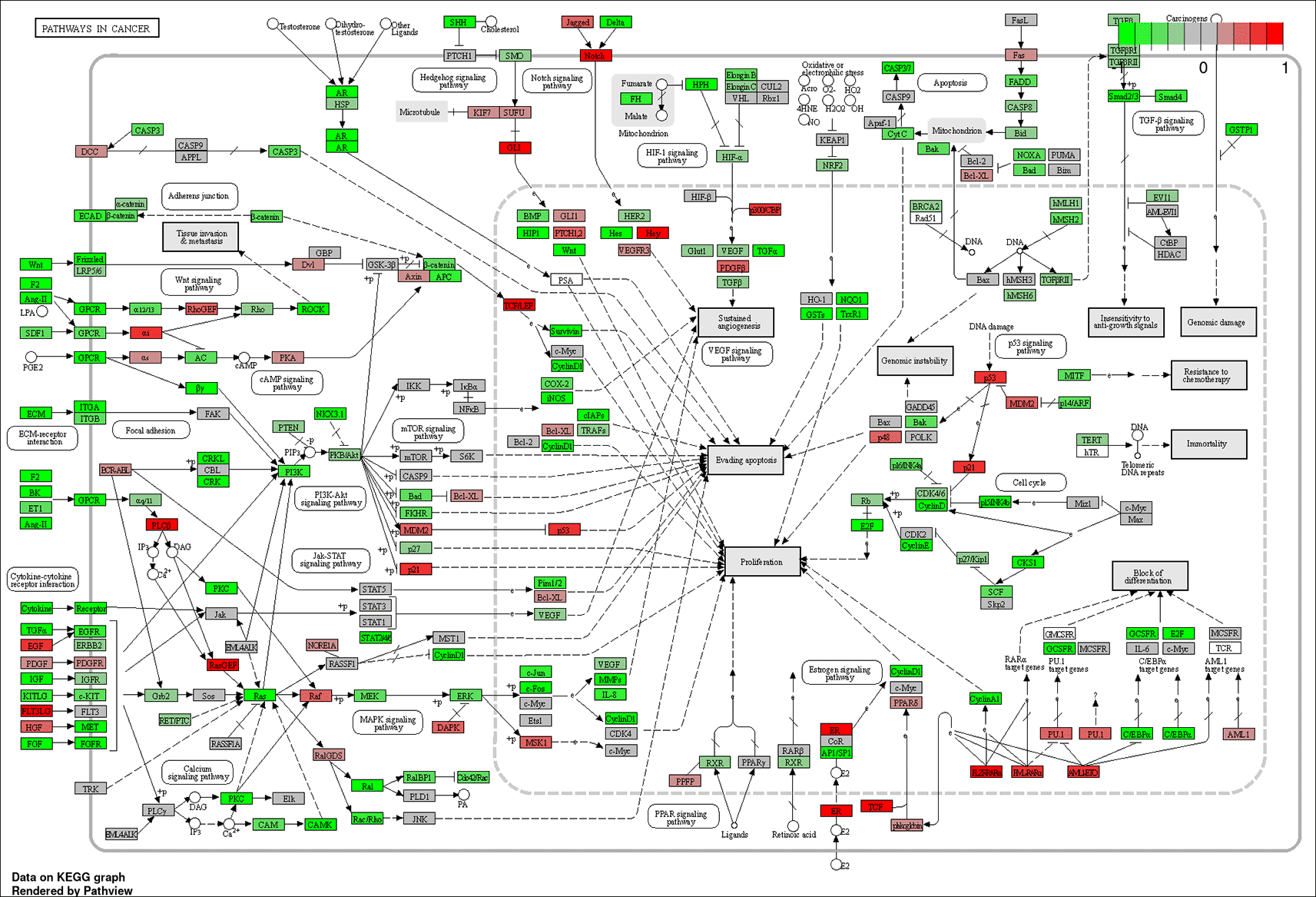
Genes colored bright red are most upregulated, and those colored bright green are most downregulated. The most significant sub-pathways within this framework appear to be: cytokine-cytokine receptor interaction, p53 signaling, extracellular membrane receptor and focal adhesion interactions, Wnt signaling, PI3K-Akt signaling, MAPK signaling, calcium signaling, cell cycle, TGF-b signaling, HIF-1 signaling, Notch and Hedgehog signaling, estrogen and androgen signaling, and block of differentiation. These pathways may provide clues regarding which molecular events confer SI-NET metastatic potential.
As mentioned in the section above, patient 0008 had several metastasis-specific splicing mutations illuminated by RNA analysis. This included a mutation in UBE4B resulting in intron retention (Figure 4), and a mutation in MXRA5 leading to activation of a cryptic splice site and loss of mRNA.
Splicing mutations were also observed in the tumors of patient 0006. He had a splice-acceptor SNV in PYGL, which encodes liver glycogen phosphorylase, in both the primary and metastatic tumor in the liver. The splicing mutation resulted in intron retention. PYGL was listed as a mutated gene in SI-NETs in a previous study (Francis et al. 2013).
Limitations of this study include its relatively small sample size, albeit for a rare tumor type, and the fact that we utilized whole exome data (thereby ignoring introns). It is possible that the molecular drivers of small bowel carcinoid tumorigenesis and progression/metastasis are in noncoding regions. Noncoding RNAs – including microRNAs (miRNAs), long noncoding RNAs (lncRNAs), small interfering RNAs (siRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), and PIWI-interacting RNAs (piRNAs) – have been shown to play a role in the pathogenesis of various tumor types (Byron et al. 2016; Zeuschner, Linxweiler, and Junker 2020). A recent study also identified differentially expressed lncRNAs in primary/metastatic SI-NET pairs (Chen et al. 2023). Additionally, given the fact that two of the mutations identified in this study were in genes involved in chromatin remodeling (i.e., ATRX and SMARCA2), it will be important to study the epigenomic landscape of SI-NETs in the future (Kidd et al. 2021; Walter et al. 2018). Proteomic analysis also appears to be informative in understanding the underlying mechanisms of carcinoid metastasis (Blažević et al. 2023). Lastly – as disparities exist in terms of carcinoid incidence, treatment, and survival – evaluation of the associations between race, ethnicity, and genetic ancestry/similarity with carcinoid genetic aberrations may be informative in a larger sample dataset (Takayanagi et al. 2022; Kessel et al. 2021).
In conclusion, we identified several candidate mutations potentially involved in the pathogenesis and metastatic cascade of SI-NET (“carcinoid”) tumors. Mutations that were identified as metastasis-specific may provide insight into intermediate steps between initial tumorigenesis and metastasis (i.e., drivers of metastatic potential). We confirmed the presence of previously reported molecular aberrations (i.e., loss of chromosome 18 and LOF mutations in CDKN1B). It remains unclear whether there is a key tumor suppressor or set of tumor suppressors on chromosome 18, the loss of which is important to the etiology of small bowel carcinoids. Gene knock-outs could be performed to determine which regions on chromosome 18 are necessary and/or sufficient to drive the formation of SI-NETs. Additionally, it is worth noting that one potential explanation that could account for the fact that previous studies identified mutations in CDKN1B in a minority of SI-NETs is the possibility that distinct molecular subtypes of small bowel carcinoids exist. The significant utility of considering transcriptomic data in addition to genomic data was exemplified by our detection of phenomena such as intron retention, splicing variants, and ASE. The addition of RNAseq data also enabled us to investigate the consequences of mutations called on the DNA level in mRNA, and perform differential expression and pathway analyses to identify several pathways potentially involved in conferring metastatic potential to SI-NETs.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)