Keywords
PDAC, pancreatic cancer, FN1, SPARC, MYL12A
This article is included in the Cell & Molecular Biology gateway.
This article is included in the Oncology gateway.
PDAC, pancreatic cancer, FN1, SPARC, MYL12A
With one of the lowest survival rates among any cancer, pancreatic cancer (PC) is a gastrointestinal malignancy with exceptional lethality. Despite its relatively low incidence rates, PC is the seventh leading cause of cancer-related death worldwide and the third in the USA after lung and colorectal cancer. The incidence rates are higher in more developed countries1 and display a rising tendency. By 2030, pancreatic cancer is predicted to be the second leading cause of cancer-related mortality in the USA.2 Approximately 90% of pancreatic cancer cases belong to pancreatic ductal adenocarcinomas (PDAC), arising from precursor lesions termed pancreatic intraepithelial neoplasia. The cause of the disease is multifactorial, but cigarette smoking appears to be one of the primary risk factors. However, alcohol consumption, obesity, aging, diabetes mellitus, and family history are significant contributors as well.3–5
Unfortunately, most patients seldom exhibit symptoms at the early stages of the disease. The wide range of non-specific symptoms, including jaundice, back- and abdominal pain, and weight loss, hampers precise diagnosis. In addition, existing screening biomarkers, including serum levels of CA19-9, display relatively low sensitivity and specificity.6 As a result, 80-85% of patients are diagnosed with unresectable tumors characterized by regional spread or metastatic disease. Only 24% of diagnosed patients survive their first year, while five-year survival is as low as 10%. Currently, surgery is the only available potentially curative option.7 Based on the extent of the disease, patients are allocated into one of four categories: resectable, borderline resectable, locally advanced, and metastatic. Altogether, only 15-20% of patients are good candidates for pancreatectomy, and the five-year survival after surgery remains low at 25%. Adjuvant therapy after surgery, including gemcitabine plus capecitabine, or modified FOLFIRINOX (oxaliplatin, irinotecan, leucovorin, and fluorouracil), is the preferred option, as both treatment regimens improve overall survival.8,9 The combination therapies based on modified FOLFIRINOX, or gemcitabine with nab-paclitaxel for metastatic pancreatic cancer, became the gold standard.10–12 In the recent PRODIGE24-trial, a modified FOLFIRINOX regimen resulted in prolonged disease-free (21.6 vs. 12.8 months) and overall survival (54.4 vs. 35.0 months) compared with gemcitabine monotherapy. Nearly 20% of FOLFIRINOX treated patients were alive at 18 months compared to only 6% of those treated with gemcitabine alone.9 Locally advanced disease with vascular involvement is regularly treated with systemic chemotherapies approved in the metastatic setting. Despite standard chemotherapy, the vast majority of resected patients experience recurrence and die within a short period.
Despite extensive molecular characterization, personalized treatment strategies of PDCA are still limited. About 5-9% of pancreatic cancer patients carry germline BRCA1 or BRCA2 mutations. In 2019, the FDA approved olaparib, a PARP-inhibitor, as maintenance therapy for metastatic patients with BRCA1/BRCA2 mutations who had not progressed during first-line platinum-based chemotherapy. Olaparib therapy improved progression-free survival in this setting compared to placebo and became the first approved targeted therapy for pancreatic cancer.13 Other approaches, including the adoption of immune-checkpoint inhibitors, did not yet translate into clinical benefits, as pancreatic tumors are characterized by highly immuno-suppressive microenvironments.14 The diverse response to treatment sparked initiatives for patient classifications, but classical histopathological features lack clinical utility.15 Based on the further genome-wide screening of transcriptional changes, PDCA has been classified into two,16 three,17 or four18 distinct molecular subtypes. Cross-comparison of these studies revealed reproducible biologic subgroups with some prognostic relevance, but the described molecular subtypes have not yet been validated or translated into successful therapeutic decisions.19 Nevertheless, principal drivers of pancreatic tumorigenesis have been identified, including KRAS, SMAD4, CDKN2A, and TP53.1,20,21 Because so few patients can have a successful surgery, and the number of therapies is limited, identifying novel potential biomarkers for early diagnosis and targeted interventions are of utmost importance.
In this study, our aim was to exploit gene expression alterations between normal and tumor tissue samples. However, instead of searching for genes upregulated in tumors, we approached biomarker selection from a novel perspective. In normal, diploid tissue samples, gene co-expression correlations frequently signal shared biological functions with coordinated regulation. On the other hand, malignant tumors frequently undergo abnormal genetic transformations, but some correlations remain consistent across normal tissues and tumor samples. We speculated that maintained correlations might be vital for cellular proliferation and survival; thus, identifying preserved correlations in gene expression across normal and tumor tissue samples can provide new targets for developing alternate therapies.
We searched the NCBI GEO database (http://www.pubmed.com/geo) for raw microarray gene chip data focusing only on Affymetrix Human Genome U133A HGU133_2 array platforms. Out of the identified 19 datasets, studies based on cell lines and xenograft models were excluded. We have also removed experiments based on pooled samples. Duplicate samples were filtered by examining the expression profiles of the first 20 genes in each sample. In case of duplications, the first published version was retained. Samples with poor description and missing raw data were rejected.
The raw CEL files were MAS5 normalized using the Affy Bioconductor library in the R statistical environment (http://www.r-project.org).22 To reduce the batch effect, a second scaling normalization was performed to set the mean expression on each chip to 1000.23 The appropriate probe set for each gene was selected with the JetSet annotation and correction package.24 We integrated the established gene expression dataset into our previously established TNMplot.com platform,25 allowing future exploitation of the database.
We searched for RNA-seq-based studies from the TCGA project at the Genomic Data Commons (GDC) portal (https://portal.gdc.cancer.gov/) and the Genotype-Tissue Expression (GTEx) portal. Data from primary tumors and normal samples were obtained from the TCGA, while RNA-seq data from normal pancreatic tissue data were obtained from GTEx. Raw counts were normalized by the DESeq2 R package, after which a second scaling normalization was performed.26
Statistical analysis and data processing were performed within the R software package (version 4.0.4). Genes with median expression below the cutoff value of 500 were excluded from subsequent analyses to reduce the chance of noise discoveries. Expression differences for each gene between normal and tumor tissues were compared with the Mann-Whitney U test. Statistical significance was accepted at p < 0.01. We used the Benjamini-Hochberg method to correct for multiple comparisons by calculating the false discovery rate (FDR). Pearson correlation analysis was performed on genes common across the two datasets. A Kruskal-Wallis test was conducted to compare gene expression across normal, tumor, and metastatic tissue samples. The ggplot2 R package was used for data visualization.27 For selected genes, survival analysis was performed by Cox proportional hazards regression using all cutoff values between the lower and upper quartiles of expression. In addition, Kaplan-Meier plots were drawn to visualize survival differences. Only genes performing at FDR ≤ 10% were considered significant for selecting the most robust biomarker candidates.
For the determination of functionally associated genes, we used the clusterProfiler28 R package and Enrichr, an open-source web-based gene enrichment analysis tool (https://maayanlab.cloud/Enrichr/), that integrates results from multiple libraries.29 Within Enrichr, the list of pathways related to our gene list was determined with the KEGG pathways option. Pathways with a Fisher exact test p-value < 0.05 and high combined score were selected. Biological processes associated with the selected list of genes were determined by GO Biological Process function. Only the top 20 significant terms were considered. The listed q-values are based on adjusted p-values corrected for multiple hypothesis testing using the Benjamini-Hochberg method. We used the CytoScape platform30 and the built-in stringApp31 for network analysis.
The combined microarray (GEO) dataset consists of 248 pancreatic tumors and 108 normal tissue samples, allowing the analysis of 12,210 genes. The RNA-seq dataset incorporates 177 tumors and four normal samples from TCGA, and 248 normal tissue samples from GTEx, enabling the analysis of 21,479 genes.
Each dataset underwent multistep filtering: gene expression differences were assessed between tumor and normal tissue samples based on log2FC values. Genes with log2FC > 0.5 or log2FC < -0.5 from the GEO dataset, and genes with log2FC > 1 or log2FC < -1 from the RNA-seq dataset were incorporated into subsequent analyses. Altogether 6088 genes had altered expression at p < 0.01 across tumor and normal tissue samples in the GEO dataset, while 17,967 genes at p < 0.01 in the RNA-seq dataset. Genes with median expression below the cutoff value of 500 were excluded from subsequent analyses to reduce the chance of noise discoveries; therefore, the number of genes was reduced to n = 2,205 in the GEO dataset and n = 3,773 in the RNA-seq dataset.
We identified 309 common genes in both RNA-seq and microarray datasets with altered gene expression across normal and tumor tissues (Figure 1A). In general, more overexpressed genes were identified in tumors compared to normal tissues. Out of the 309 genes, 267 had increased expression in primary tumors compared to 42 genes overexpressed in normal samples, both in GEO and RNA-seq datasets. Genes with altered expression across normal and tumor tissues are graphically represented by a VOLCANO plot (Figure 1B).
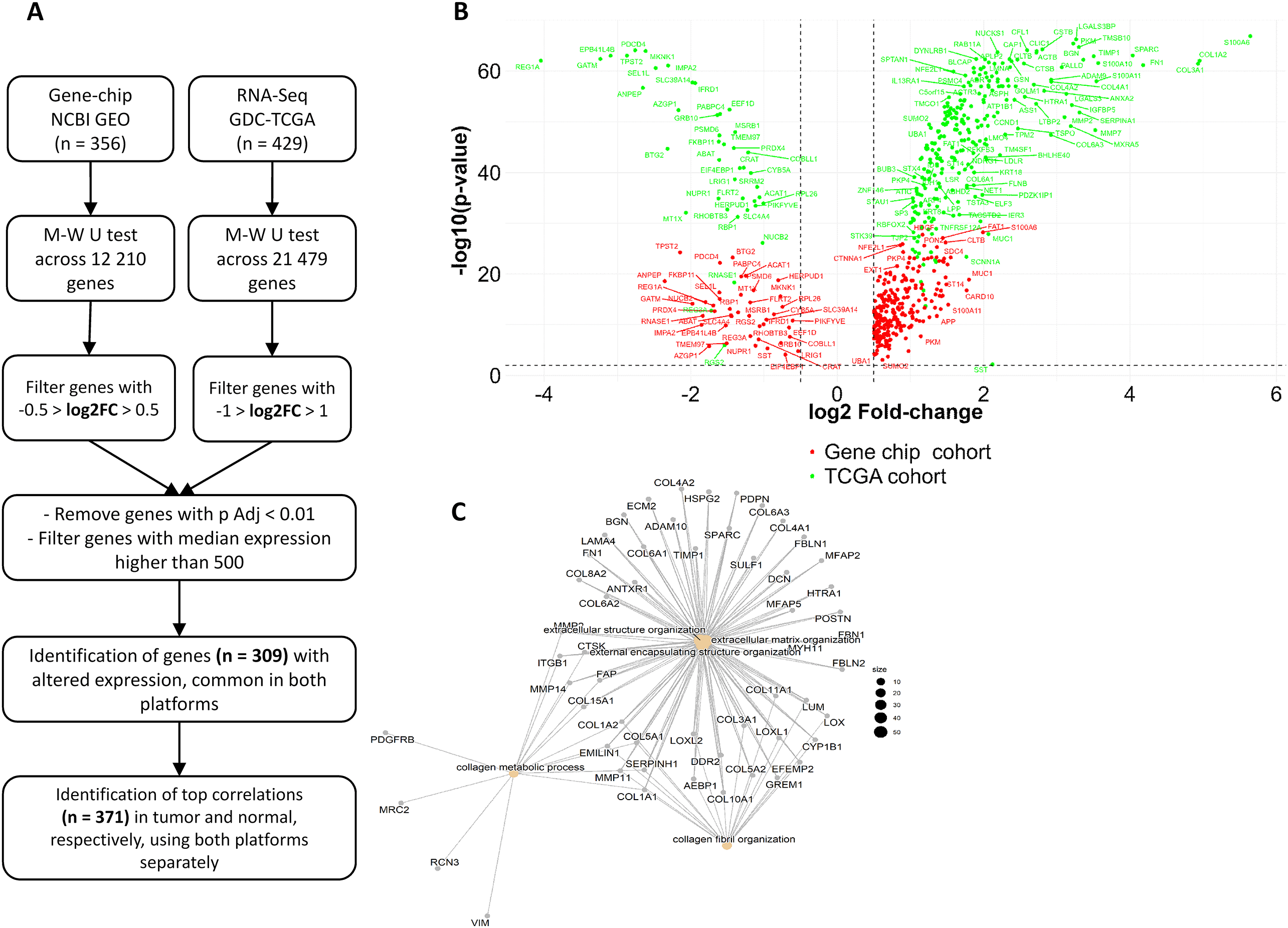
A Pearson correlation was performed to identify associations between the 309 differentially expressed genes and all genes in the gene list. We analyzed correlations in tumors and normal tissue samples separately, then common correlations were identified. We have filtered out correlations with a correlation coefficient below 0.5.
Altogether, we have identified 371 preserved correlations involving 262 individual genes across tumors and normal tissue samples. These preserved correlations were robust, as were independently identified in both microarray and RNA-seq datasets with R > 0.5 or R < -0.5, and p < 0.01 (Supplemental Table 1). The network of significantly correlated genes is illustrated in Figure 1C. The most significant top 20 correlations in both normal tissues and tumor samples in each dataset are presented in Table 1.
We observed differences in the strength of preserved correlations between normal tissues and tumor samples. In normal samples, the strongest correlations involved mainly genes encoding collagens both in RNAseq and GEO data. Conversely, in tumors, we observed a shift in the strength of correlations. Genes associated with the regulation of focal adhesion and actin cytoskeleton (ACTR3, MYH11, MYL9, MYLK) along with members of the Heat Shock Protein Family (e.g., HSPA4, HSPH1, HSPA1B), gained momentum and participated in the strongest correlations compared to normal samples (Table 1).
Seventy-nine genes from the identified 309 differentially expressed genes participated in preserved correlations, out of which 73 genes were upregulated in tumors compared to normal tissue samples (Supplemental Table 2).
Some genes overexpressed in tumor samples were strikingly overrepresented among the 371 preserved correlations, including SPARC, COL6A3, MMP2, HTRA1, FN1, PALLD, and COL3A1, some participating in as many as 58 correlations (Figure 2A). Over 70% of all preserved correlations (263 altogether) involved at least one of these seven genes.
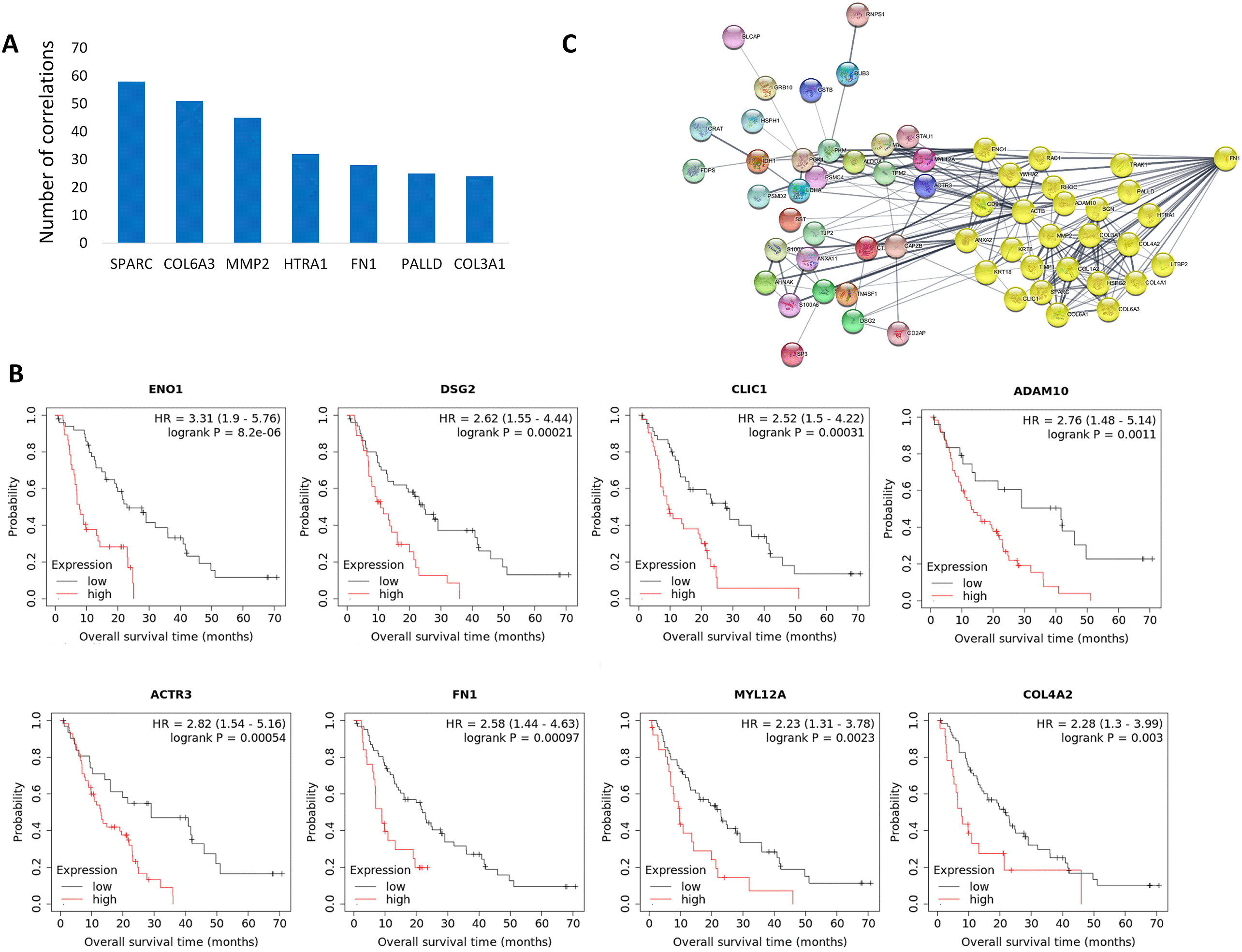
Out of the 262 genes participating in 371 preserved correlations, high expression of 28 genes was associated with worse overall survival at FDR 10%. Out of these, 16 genes (DSG2, FN1, ACTR3, PKM, CAPZB, TSC22D1, COL4A2, ADAM10, ENO1, CLIC1, PARP12, PPP4R1, PSMC4, KRT18, MYL12A, and PSMD2) were upregulated in tumors compared to normal samples (Table 2, Figure 2B).
Out of these 16 genes were upregulated in tumors compared to normal tissue samples. Genes in the boldface were overexpressed in tumors.
Among the upregulated genes the expression of ENO1 (p = 8.2E-06), DSG2 (p = 0.00021), CLIC1 (p = 0.00031) and ACTR3 (p = 0.00054) showed a particularly strong association with disease outcome (FDR ≤ 2%) (Figure 2B). The heavily overrepresented FN1 gene was also associated with worse overall survival at FDR 5% (p = 0.00097), providing promising biomarker candidates for subsequent clinical validations. According to a String functional network analysis, the FN1 protein has a central role in the associated protein network of upregulated genes (Figure 2C).
The 371 preserved correlations incorporated 79 of the initially identified differentially expressed genes, out of which the majority, 73 genes, were overexpressed in tumors compared to normal tissues. With the help of TNMplot.com, we have also compared the expression pattern of these genes between primary tumors and metastases. The expression of MYL12A increased most significantly in metastatic samples compared to primary tumors (Figure 3A). Of note, the expression of both MYL12A and MYL12B gradually intensified across normal samples, primary tumors, and metastatic tissue samples (MYL12A, KW p = 2.57E-11; MYL12B KW p = 1.38E-14). We have observed a similar pattern in seven additional genes, including ENO1 and CLIC1, that were previously identified as robust prognostic biomarkers of survival (Figure 3B).
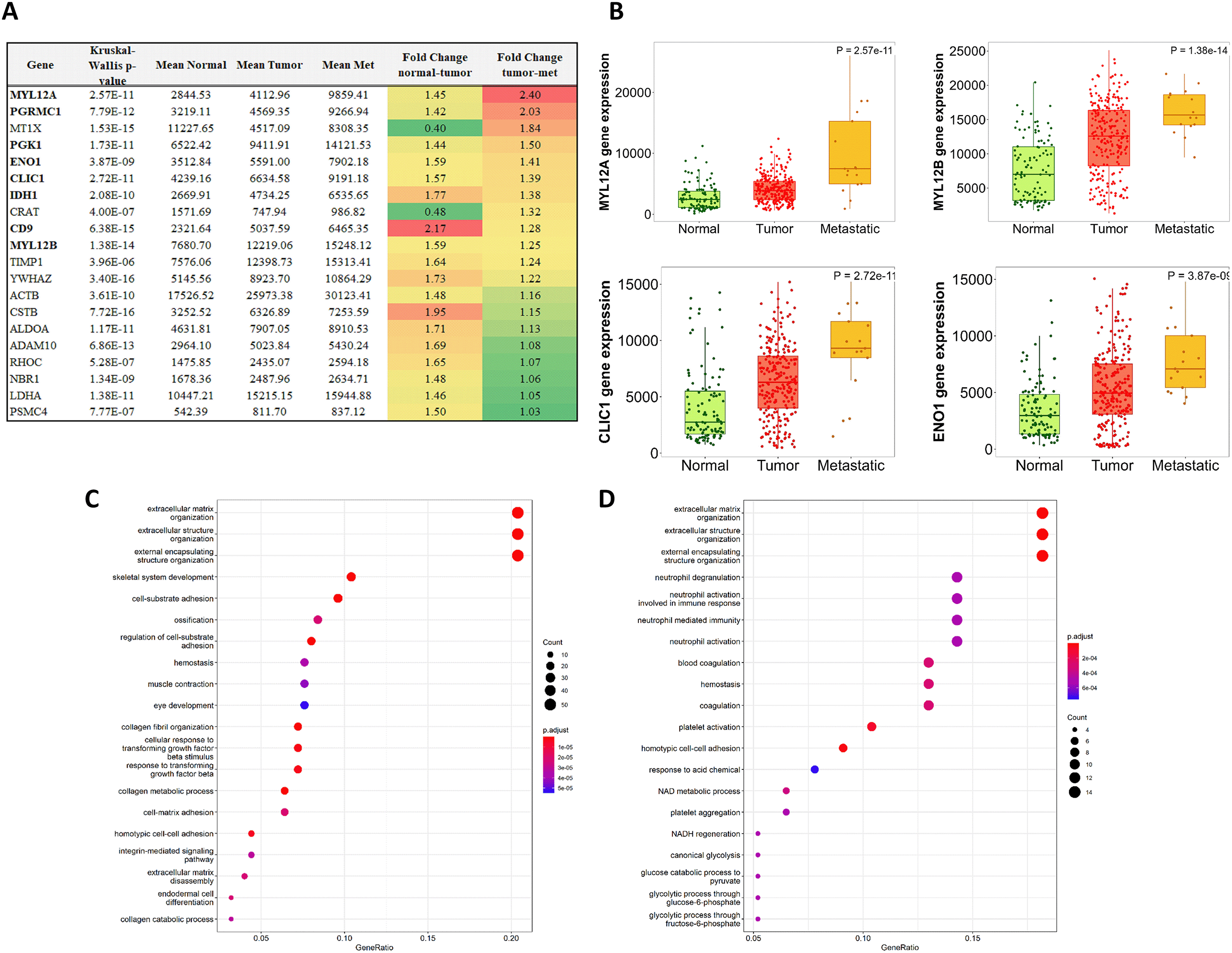
Expression differences are color-coded: red encodes increasing expression from normal to tumor and from tumors to metastatic samples, while green encodes decreasing expression (A). The expression of MYL12A, MYL12B, CLIC1 and ENO1 genes gradually increased from normal tissue samples to primary tumors and metastatic samples (B).
The close-knit network of 262 genes involved in preserved correlations is mainly associated with ECM organization and function (C). Gene ontology analysis involving the 79 differentially expressed genes from preserved correlations revealed similar top functions (D).
We subjected the 262 genes from preserved correlations to gene enrichment analysis. The top biological functions were Extracellular matrix organization, Extracellular structure organization, and External encapsulating structure organization (Figure 3C). The most significantly enriched terms based on KEGG Pathway analysis were Focal adhesion, Protein digestion and absorption, ECM-Receptor interaction, Tight junction, Proteoglycans in cancer, regulation of actin cytoskeleton, and AGE-RAGE signaling pathway in diabetic complications (Table 3A). According to the GO Cellular compartment analysis in Enrichr, 54 genes were significantly associated with the collagen-containing ECM (Supplemental Table 3).
Top functions remained similar in both cases.
The ITGB1, COL1A2, COL4A1, COL4A2, and FN1 genes were predicted to be involved in multiple cancer-related pathways, such as ECM-receptor interactions, the PI3K-Akt signaling pathway, human papillomavirus infection, and small cell lung cancer.
When we narrowed down the gene enrichment analysis to the 73 genes overexpressed in tumors participating in preserved correlations, the most significantly enriched biological functions and KEGG pathways remained similar (Table 3B, Figure 3D).
Pancreatic cancer is one of the most lethal malignancies with limited options of therapies and biomarkers. In order to pinpoint survival-specific vulnerabilities, we aimed to identify upregulated genes with preserved correlations across normal and tumor samples. Previous research revealed profoundly different biomolecular networks underlying cancer conditions compared to healthy tissues.32 Notwithstanding, we speculated that sustained correlations might offer insight into fundamental biological functions, providing potential novel targets for therapeutic interventions. To increase robustness, we collected data from two independent platforms, including RNA-seq and microarray-based studies. Then, we identified genes with an altered expression between tumors and normal tissues and looked for correlations preserved across both data platforms.
We have identified 371 preserved correlations comprising 262 genes, including 79 genes with altered expression across tumors and normal samples. These preserved correlations were robust, as were independently identified in both microarray and RNA-seq datasets. Moreover, a few of these differentially expressed genes remarkably dominated preserved correlations, including SPARC, COL6A3, COL3A1, MMP2, HTRA1, FN1, and PALLD. Altogether, 16 genes upregulated in tumor tissues were linked to dismal disease outcomes, out of which the expression of MYL12A gradually increased during disease progression.
Upon close examination, most of these genes are associated with the tumor microenvironment, participating in extracellular matrix (ECM) organization. PDAC has a dense fibrotic stroma, composed of abundant extracellular matrix and other non-neoplastic cell types, responsible for chemo- and radiotherapeutic resistance.33 The abundant ECM deposition, called desmoplastic reaction, plays a crucial role in PDAC. Beyond structural support, the ECM functions as a reservoir of growth factors and a mediator of complex signaling pathways.34,35 The loss of ECM homeostasis and integrity is one of the hallmarks of cancer, resulting in cancer progression36; thus, potential targeting and modifications of the ECM in PDAC are being intensely studied both in the preclinical and clinical settings.37,38
Our results corroborate the importance of ECM organization in disease outcomes. Most importantly, we pinpoint critical interactions as potential targets for combination therapies. Fibronectin (FN1) is a principal constituent of the extracellular matrix that provides the scaffold to build up the entire ECM structure, affecting its architecture and composition. It is overexpressed in PDAC as part of the desmoplastic reaction, is associated with aggressive tumor characteristics,39 and functions as a primary driver of tumor progression by enhancing growth factor signaling.40 FN1 secreted by pancreatic stromal cells promoted gemcitabine resistance by downregulation of ERK1/2 phosphorylation.41 A synthetic FN1 blocking-peptide (Arg-Gly-Asp-Ser) abrogated the gemcitabine resistance in PDAC cell lines.41
Our datasets identified both FN1 and the matricellular protein SPARC remarkably overrepresented in preserved correlations across normal and tumor samples. High stromal expression of SPARC was linked to poor patient prognosis in PDAC,42 and pancreatic stellate–cell-derived SPARC promoted proliferation of PDAC cells in vitro.43 Recent research indicated that the presence of plasma fibronectin determines the specific effect of SPARC on pancreatic cancer cells: depletion of fibronectin switched SPARC from promoting cancer cell proliferation to growth inhibition and induction of apoptosis.43 In our data both FN1 and SPARC expression were correlated with MMP2 in normal and cancer tissue samples. MMP2 is a member of the matrix metalloproteinase (MMP) gene family, primarily located in the tumor stroma, capable of cleaving collagens and other components of the extracellular matrix involved in signal transduction.44 In PDAC, MMP2 is among the most studied gelatinases, with a potential clinical relevance45: MMP2 expression is linked to the activity and the invasive potential of pancreatic cancer cell lines, also associated with the development of the characteristic desmoplastic reaction.46 The potential importance of MMP2 in PDAC progression is supported by studies using selective MMP2, MMP9, and MMP14 inhibitors in mice and Syrian hamsters, such as MMI-166, RO28-2653, OPB-3206, and SB-3CT.45
The most abundant component of the mammalian ECM is collagen. Collagen Type IV Alpha 2 Chain (COL4A2) encodes one of the six subunits of collagen type IV, a major structural component of basal membranes.47 In our dataset, COL4A2 expression was highly correlated with COL4A1, another type IV collagen alpha protein found in most connective tissues. In pancreatic cell lines and xenograft models of pancreatic cancer, type IV collagen production exceeded that of other ECM proteins examined, including collagen type I.48 The abundantly produced collagen type IV is deposited near the pancreatic tumor cells, creating a basal membrane-like structure.49 The structure colocalizes with integrin receptors on the cancer cell surface, stimulating the proliferation and migration of pancreatic cancer cells. Thus, type IV collagens provide essential survival signals to the pancreatic cancer cells through an autocrine loop.49 High circulating level of postoperative collagen IV has been linked to dramatically reduced survival in PDAC patients undergoing curative surgery.50
Substantiating previous findings, we found an association between high COL4A2 expression and poor prognosis, supporting the role of COL4A2 as a potential prognostic biomarker of PDAC. The drug candidate T12, a rationally designed inhibitor of collagen IV network formation, reduced both COL4A1 and COL4A2 expression and disrupted EMT-based chemo-resistance in mouse models of breast and lung cancers without apparent toxicity.51 Thus, targeting specific EMC proteins, including COL4A2 and COL4A1, may represent a novel therapeutic opportunity for treating PDAC.
Another protease, the ADAM Metallopeptidase Domain 10 (ADAM10), has also been heavily investigated by pharmaceutical companies. ADAM10 is ubiquitously expressed and cleaves many substrates involved in cancer signaling, such as PD-L1, NOTCH, EGFR/HER-ligands, among others.52 The evidence for ADAM10 as a suitable cancer biomarker increases: ADAM10 overexpression correlates with poor patient prognosis in cancers of the colon, lung, ovary, uterus, and stomach.53 Moreover, it has a substantial value as a prognostic biomarker in glioblastomas, sacral chordomas, and triple-negative breast cancer and became a potential target.52,54,55 In PDAC, ADAM10 was suggested to be a driver of tumor progression and fibrosis after radiation therapy. Pharmacological inhibition of ADAM10 delayed fibrosis and improved survival in radiation therapy-treated orthotopic and metastatic mouse models.56 Targeting the active ADAM10 with a monoclonal antibody, such as mAb 8C7, that masks the substrate-binding pocket preferentially located in tumors compared to normal tissue may offer targeted inhibition to prevent tumor growth.57
Our results suggest additional insight into the evolving tumor microenvironment during disease progression. The expression of MYL12A was associated with poor prognosis and correlated with MYL12B upregulation within both normal and tumor samples. The expression of both genes increased during tumor progression, from normal tissues to primary tumors to metastatic samples. Myosin Light Chain 12A (MYL12A) and 12B (MYL12B) regulate smooth muscle and non-muscle cell contractile activity. Both genes are implicated in cytokinesis and cell locomotion,58 and intensifying expression levels indicate the acquisition of motility by cancer cells. The biological functions and rising expressions during cancer progression warrant the role of MYL12A both as a potential prognostic biomarker and a therapeutic target of PDAC.
We observed an increase in the expression of ENO1 and CLIC1 genes between normal samples and tumor tissue. Moreover, in our data, overexpression of both ENO1 and CLIC1 was associated with poor overall survival. Alpha enolase (ENO1) is a glycolytic enzyme localized on the cell surface with a complex role in cancer progression: ENO1 promotes glycolysis and ECM degradation, affects actin remodeling, facilitates tumor invasion, and migration, and activates cancer signaling pathways.59 During inflammation, enolase migrates to the cell surface that promotes the production of pro-inflammatory cytokines, chemokines, reactive oxygen species, and nitric oxide.60
Higher ENO1 expression has been associated with larger tumor size, increased invasion, shorter survival, and poor prognosis in several solid tumors, including hepatocellular carcinomas, breast- and lung cancers,59 and PDAC.61 ENO1 has clinicopathological and diagnostic significance as a tumor-associated antigen as well: combining ENO1 autoantibodies with other biomarkers improved diagnostic sensitivity and accuracy of lung cancer detection.62,63 In PDAC, several fundamental characteristics designate ENO1 to be an ideal biomarker candidate: it is localized on a cell surface, making it targetable; it is upregulated in tumors compared to normal tissues, is associated with clinical outcome, and has vital metabolic functions. Small molecule inhibitors (e.g., ENOblock, ENO1i) are already being investigated in various preclinical settings and disease models, including spinal cord injuries and multiple myelomas.64,65 Additionally, DNA vaccinations targeting ENO1 in combination with chemotherapy may offer promising alternatives in PDAC treatment. In pancreatic cancer-prone mice, treatment with gemcitabine combined with anti-ENO1 vaccine elicited a robust antitumor response and reduced the size of PDAC lesions.66 Administration of an anti-ENO1 mAb inhibited in vitro invasion of human PDAC cells and their metastatic spread in immunocompromised mice; thus, targeting ENO1 may be exploited as a novel therapy to increase the survival of metastatic PDAC patients.67
PDAC presents unique challenges for chemotherapy and immunotherapy with an immunosuppressive microenvironment and a dense stroma, creating a physical barrier to drug delivery; thus, alternative therapeutic targets, such as ion channels, provide an increasingly evaluated option. Ion channels are involved in every aspect of oncogenesis, influencing critical processes within each hallmark of cancer. Chloride Intracellular Channel 1 (CLIC1) plays a vital role in many physiological functions, including ion homeostasis, acidity, and cellular volume, considered to be both the sensor and executor of oxidative stress. It can exist in a soluble form, but in pathological states, CLIC1 is expressed explicitly as a transmembrane chloride channel.68 CLIC1 overexpression has been established in pancreatic cancer tissue and is indicated to be a putative oncogene.69–71
In summary, our findings provide novel insight into intimate gene-networks corroborating previous knowledge about interactions within the tumor microenvironment. Although our results are based on in silico approaches, maintained correlations across RNA-seq and gene chip platforms substantiate the significance of our findings. Our results may be utilized to develop combination therapies against PDAC and offer novel ideas to be explored in future studies.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)