Keywords
Anuran, genome assembly, transcriptome assembly, mitogenome, reference genome, Myobatrachidae
This article is included in the Genomics and Genetics gateway.
Anuran, genome assembly, transcriptome assembly, mitogenome, reference genome, Myobatrachidae
In this revision, we add context for the discrepancy between the short-read estimation of genome size (3.1 Gb) and the assembled genome size (5.519 Gb). We hypothesise that the underestimation of genome size by short-read data was due to known limitations of short-read assemblies of repetitive regions of the genome. The Kroombit tinkerfrog genome was highly repetitive, with 63.35% of the total sequence annotated as repeat elements. We have also updated Figure 2 to be more easily readable. No other changes have been made.
See the authors' detailed response to the review by Natalie Forsdick
See the authors' detailed response to the review by Sandra Goutte
The Kroombit tinkerfrog (Taudactylus pleione) is a stream-dwelling Anuran of the Myobatrachidae family. It is endemic to Queensland, Australia, with a distribution restricted to fragmented patches above 400m altitude on an isolated plateau in the Kroombit Tops temperate rainforest (Skerratt et al., 2016). The Kroombit tinkerfrog is listed as Critically Endangered by the International Union for the Conservation of Nature (IUCN) with less than 200 individuals estimated to remain in just a 19 km2 area of occupancy (IUCN SSC Amphibian Specialist Group, 2022) and is the highest ranked frog species requiring management action in Australia (Gillespie et al., 2020). Threatening processes include infection by chytrid fungus Batrachochytrium dendrobatidis, habitat degradation due to agriculture, feral animals and plants, and fire (Hines, 2014). The Kroombit tinkerfrog was identified as one of seven Australian amphibians at high risk of extinction due to chytridiomycosis (Skerratt et al., 2016), and the fifth most likely frog to go extinct in an analysis of 26 Critically Endangered and Endangered Australian frogs (Geyle et al., 2022). A captive breeding program was established at Currumbin Wildlife Sanctuary in 2018, with the aim of releasing captive-bred tinkerfrogs back to the wild.
The Taudactylus genus is estimated to have diverged from other myobatrachids 65 million years ago, contributing to the high Evolutionary Distinctiveness and Global Endangerment (EDGE) score of 6.52 for the Kroombit tinkerfrog, which places it as the seventh highest EDGE amphibian (Zoological Society of London, 2020). However, there are currently no published reference genomes available for the Taudactylus genus. The Kroombit tinkerfrog is primarily nocturnal and is secretive, making it difficult to find (Clarke, 2006). Characterising the mitochondrial genome may therefore assist in efforts to develop environmental DNA (eDNA) approaches for monitoring the species in the wild using freshwater samples, as has been demonstrated in other endangered frog species (Eiler et al., 2018; Villacorta-Rath et al., 2021). Therefore, in this study we sequenced DNA and RNA to assemble the genome, mitogenome, and transcriptomes and provide the first genomic resources for the Kroombit tinkerfrog.
Due to the critically endangered status of the Kroombit tinkerfrog, we did not lethally sample an adult. Instead, three tadpoles of unknown sex from the captive breeding program at Currumbin Wildlife Sanctuary were medically euthanised due to a failure to thrive, by immersion in 10 mL of 250 mg/L Tricaine MS222, buffered to pH 7 with sodium bicarbonate until cessation of a visibly detectable heartbeat, or in very small tadpoles, an absence of reflexes after prolonged immersion (University of Sydney Animal Research Authority 2021/1899). Tadpoles were then either flash frozen at -80°C or preserved in RNALater before being stored at -80°C. The tadpoles were skinned to avoid pigmentation issues that could impact sequencing. High molecular weight (HMW) DNA was extracted from the flash frozen tadpole tissue using the Nanobind Tissue Big DNA Kit v1.0 11/19 (Circulomics). A Qubit fluorometer was used to assess the concentration of DNA with the Qubit dsDNA BR assay kit (Thermo Fisher Scientific). RNA was extracted from the other two tadpoles preserved in RNALater, using the RNeasy Plus Mini Kit (Qiagen) with RNAse-free DNAse (Qiagen) digestion. Extractions were performed using tissue from the head, midsection, and tail of the tadpoles. Only tissues from one tadpole yielded acceptable quality RNA as determined by NanoDrop (Thermo Fisher Scientific), so were sequenced.
We first performed short-read sequencing to provide an estimate of genome size, which was previously unknown. HMW DNA underwent PCR-Free DNA Preparation and Illumina NovaSeq 150-bp paired end sequencing at the Australian Genome Research Facility, Melbourne, Australia. GenomeScope v1.0 (Vurture et al., 2017) estimated the haploid genome size at 3.1 Gb. As a result, HMW DNA was sent for PacBio HiFi library preparation with Pippin Prep and sequencing on three single molecule real-time (SMRT) cells of the PacBio Sequel II (Australian Genome Research Facility, Brisbane, Australia). Additional HMW DNA from the same tadpole was later sent for sequencing on a fourth SMRT cell after the initial assembly resulted in low coverage due to a larger than expected genome (see Results).
Total RNA from the head, midsection, and tail of one tadpole was sequenced as 100 bp paired-end reads using Illumina NovaSeq 6000 with Illumina Stranded mRNA library preparation at the Ramaciotti Centre for Genomics (University of New South Wales, Sydney, Australia).
Genome assembly was conducted on an Amazon Web Services r5.24x large cloud machine (96 vCPU; 1 TB RAM). The raw circular consensus sequence reads were filtered to retain HiFi reads (≥Q20) with BamTools v2.5.1 (Barnett et al., 2011). SamTools v1.15 (Danecek et al., 2021) bam2fq converted the BAM files to FASTQ format for input to Hifiasm v0.16.1-r375 (Cheng et al., 2021, 2022). The Hifiasm assembly included the following modified parameters: -f38 (recommended for genomes larger than the human genome), -a 6 (to increase number of assembly graph cleaning rounds from the default of 4), and -s 0.65 (to reduce the similarity threshold for duplicate haplotigs to be purged).
Basic genome assembly statistics were calculated using the ‘stats.sh’ script from BBMap v38.86 (Bushnell, 2022). Completeness was assessed using Benchmarking Universal Single-Copy Orthologues (BUSCO) v5.2.2 (Simão et al., 2015) with the vertebrata_odb10 lineage (n=3,354 BUSCOs) run on Galaxy Australia (The Galaxy Community, 2022). The repetitive elements of the genome were identified and classified by building a custom database using RepeatModeler v2.0.1 (Flynn et al., 2020) and RepeatMasker v4.0.9 (Smit et al., 2013-2015) with the -nolow parameter to avoid masking of simple low-complexity repeats.
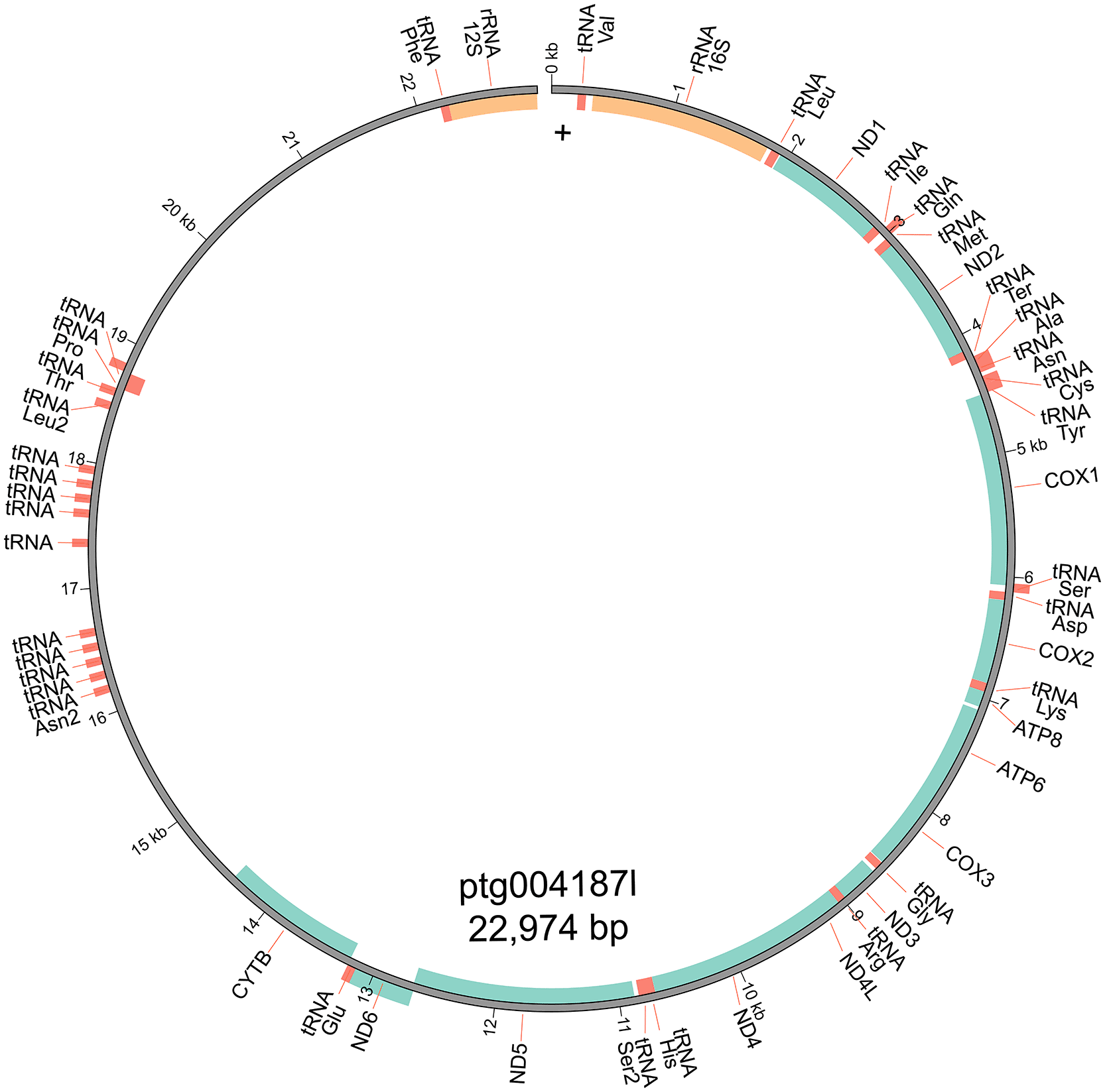
The mitochondrial genome was assembled from the genome assembly using MitoHiFi v2 (Allio et al., 2020; Uliano-Silva et al., 2023). MitoHiFi first identifies the most closely related publicly available mitochondrial genome for a similarity-based approach, in this case the Wokan cannibal frog Lechriodus melanopyga (NCBI reference sequence NC_019999.1; (Irisarri et al., 2012)). The mitochondrial genome was visualised with MitoZ v2.3 (Meng et al., 2019).
Transcriptome assembly was conducted on the University of Sydney High Performance Computer, Artemis. The raw transcriptome reads were quality assessed both prior to and after quality trimming with FastQC v0.11.8 (Andrews, 2010). Trimmomatic v0.39 (Bolger et al., 2014) was used to quality trim reads specifying TruSeq3-PE adapters, SLIDINGWINDOW:4:5, LEADING:5, TRAILING:5 and MINLEN:25. The repeat-masked genome was indexed and reads aligned with HiSat2 v2.1.0 (Kim et al., 2019). Resulting SAM files were converted to a coordinate-sorted BAM format with SamTools v1.9 view and sort. StringTie v2.1.6 (Pertea et al., 2015) generated a GTF for each transcriptome. The aligned RNAseq reads were then merged into transcripts and filtered to remove transcripts found in only one tissue with FPKM < 0.1, using TAMA-merge v2020/12/17 (Kuo et al., 2020) and CPC2 v2019-11-19 (Kang et al., 2017). TransDecoder v2.0.1 (Haas, 2022) was used to predict open reading frames in the resulting global transcriptome. The completeness of the global transcriptome was assessed using BUSCO v5.2.2 in ‘transcriptome’ mode with the vertebrata_odb10 lineage.
Genome annotation was performed using FGENESH++ v7.2.2 (Softberry; (Solovyev et al., 2006)) on a Pawsey Supercomputing Centre Nimbus cloud machine (256 GB RAM, 64 vCPU, 3 TB storage) using the longest open reading frame predicted from the global transcriptome, non-mammalian settings, and optimised parameters supplied with the Xenopus (generic) gene-finding matrix. BUSCO v5.2.2 in ‘protein’ mode was used to assess the completeness of the annotation with the vertebrata_odb10 lineage. The ‘genestats’ script (GitHub) was used to obtain the average number of exons and introns, and average exon and intron length.
Initial genome size prediction from the short-read sequencing data predicted a total haploid length of 3.1 Gb (Figure 1). The initial genome assembly using PacBio HiFi data from three SMRT cells yielded a genome of 5.59 Gb in length, comprising 9,966 contigs with a contig N50 of 2.401 Mb. We hypothesise that the short-read data underestimated genome size due to the highly repetitive nature of large amphibian genomes (Kosch et al., 2023) and the known limitations of short reads that are too short to span long repeats or may collapse repeats in the assembly (Wang et al., 2021). Coverage of the initial genome assembly was low (14×) due to the underestimation of the genome size, so re-assembly with the addition of a fourth SMRT cell yielded a genome of 5.519 Gb, comprising 4,196 contigs and with an improved contig N50 of 8.853 Mb, and a coverage of 21× (Table 1). The mitochondrial genome was 22,974 bp long and consisted of 38 genes, including 13 protein-coding genes, 2 rRNAs, and 23 tRNAs, with a GC content of 41.89% (Figure 2).

The total length of the genome sequence was estimated at 3.119 Gb.
Over 99.98% of raw reads were retained after quality trimming. The individual tissue transcriptomes had high mapping rates to the repeat-masked genome (82.8% head; 91.3% mid-section; 84.9% tail). The global transcriptome had 93.8% complete BUSCOs [Single copy: 31.4%; Duplicated: 62.4%]; 3.1% fragmented BUSCOs and 3.1% missing BUSCOs. A total of 14,448 predicted genes were used as evidence for genome annotation. Repetitive elements comprised 63.35% of the total genomic sequence, with 37.53% unclassified repeats (Table 2). A total of 70,371 genes were predicted from the annotation. This is likely to be an overestimate of the true number of protein-coding genes, expected to be within the range of 20,000 to 30,000 (Sun et al., 2020), possibly due to a lack of homology-based evidence for amphibians. There was an average of 4.9 exons (SE=0.03) and 3.9 introns (SE=0.03) per putative gene, with an average exon length of 340 bp (SE=16) and an average intron length of 7,187 bp (SE=220). The annotation had 84.1% complete BUSCOs [Single copy: 81.7%; Duplicated: 2.4%]; 9.1% fragmented BUSCOs and 6.8% missing BUSCOs.
In summary, we have generated a high-quality long-read draft annotated reference genome, mitogenome, and global transcriptome for the critically endangered Kroombit tinkerfrog, providing the first genome for the Taudactylus genus.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)