Keywords
Soda lake, spatiotemporal, archaea, bacteria, physicochemical parameters, α-diversity, β-diversity
This article is included in the Bioinformatics gateway.
Soda lake, spatiotemporal, archaea, bacteria, physicochemical parameters, α-diversity, β-diversity
Most living organisms are adapted to habitats characterized by moderate temperature (10–37°C), pH (of approximately 7), salinity (0.15–0.5 M NaCl), pressure (1 atm), and adequate supply of water (Aguilar et al., 1998; Antranikian et al., 2005). However, molecular techniques such as next-generation sequencing have revealed that diverse groups of organisms thrive even in biomes previously thought to be lifeless (Canganella & Wiegel, 2011; Rampelotto, 2013). Microbial communities in ecosystems such as the hypersaline lakes of the East African rift valley survive and thrive under one or several extreme conditions and are referred to as polyextremophiles (Sorokin et al., 2014a; Urbieta et al., 2015).
The distribution and diversity of microbial communities in hypersaline lakes is mainly affected by physicochemical parameters (Tazi et al., 2014). Lake Magadi is an example of an extreme habitat characterized by high concentrations of Na+, K+, CO32–, Cl–, HCO3–, and SiO2, but low concentrations of Ca2+ and Mg2+ (Jones et al., 1998; Getenet et al., 2022). During the dry seasons, thermonatrite (Na2CO3.H2O), and halite (NaCl) precipitate by evaporative concentration (Eugester, 1971, 1980). The lake is in a region with alternating wet and dry seasons. During the dry season, when ground temperatures exceed 40°С, there is extensive evaporation (Matagi, 2004; Muruga & Anyango, 2013). Furthermore, the lake is almost entirely covered by a white layer of soda, and flooding may occur when it rains due to feeding water from the surroundings.
Despite the extreme conditions existing in the lake, it is a highly productive ecosystem with diverse microbial communities driving active nitrogen, carbon, and sulfur cycles (Jones et al., 1998; Sorokin et al., 2007). The high productivity is mainly driven by Arthrospira spp. and other cyanobacteria (Melack & Kilham, 1974; Oduor & Schagerl, 2007). Cyanobacteria in lake lagoons only form algal mats in these lakes during rainy seasons (Jones et al., 1998; Muruga & Anyango, 2013; Krenitz & Shagerl, 2016). Reports indicate that Ectothiorhodospira, an anoxygenic phototrophic halophilic bacterium also plays an essential part in primary production (Matagi, 2004; Grant, 2006). Additionally, eukaryotes such as diatomic and green algae contribute to the primary production (Matagi, 2004).
A significant number of bacteria have been isolated from extreme environments and they often demonstrate adaptations to optimal growth under the prevailing conditions (Krulwich et al., 2011; De Maayer et al., 2014; Sorokin et al., 2014b). Validly described isolates from Lake Magadi include the archaeal genera Natronobacterium and Natronococcus gen. nov. (Tindall et al., 1984) and Natronobacterium magadii, Natrialba magadii (Kamekura et al., 1997), bacterial species Spirochaeta alkalica sp. nov., Spirochaeta Africana (Zhilina et al., 1996), Tindallia magadiensis (Kevbrin et al., 1998), Halomonas magadii (Duckworth et al., 2000), Amphibacillus fermentum (renamed Pelagirhabdus fermentum) sp. nov., Amphibacillus tropicus, and Halonatronum saccharophilum (Zhilina et al., 2001), Methylonatrum kenyense (Sorokin et al., 2007), Euhalothece natronophila (Mikhodyuk et al., 2008) and Natranaerobaculum magadiense (Zavarzina et al., 2013). Edwin et al. (2019) recovered 11 isolates affiliated with the cyanobacterial orders Chroococcales, Oscillatoriales, Pleurocapsales and Nostocales. Recent studies have reported isolates affiliated to the genus Bacillus, Alkalibacterium, Staphylococcus, Micrococcus, Halomonas, and Alkalilimnicola (Kiplimo et al., 2019; Kipnyargis et al., 2022). Orwa et al. (2020) recovered several fungal isolates affiliated with 18 different genera with Aspergillus, Penicillium, Cladosporium, Phorma and Acremonium being dominant. Several studies have explored the microbial diversity in Lake Magadi using amplicons analysis targeting groups such as fungi (Kambura et al., 2016; Salano et al., 2017; Mwirichia, 2022) or bacteria (Kambura et al., 2016).
A key ecological question is how microbial diversity changes with the fluctuating physicochemical conditions with seasons. We hypothesized that microbial communities within the lake shift in response to changes in the water chemistry over time. We predict that the communities in the brines are different from those in the open lake water. In this study, we explored the spatiotemporal variation in the microbial community over four months at different sites in Lake Magadi using Illumina sequencing.
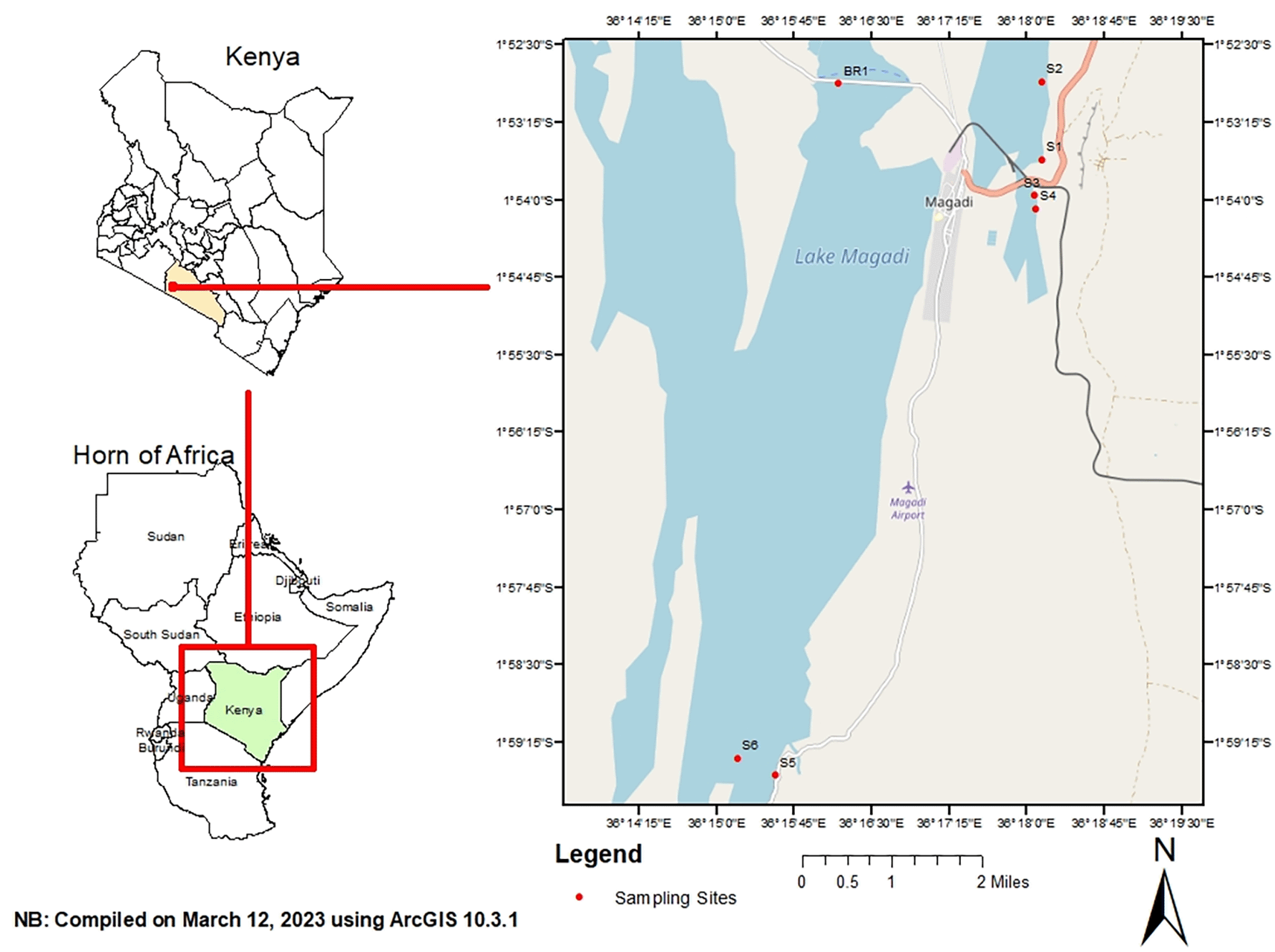
Sampling was done in hypersaline Lake Magadi, Kenya. It is located 1°43-2°00 S and 36°13-36°18E in an enclosed basin with an annual precipitation of 500 mm (Behr & Röhricht, 2000). Lake Magadi is a relatively shallow water body that is fed by various springs distributed along the edges of the lake. The inflows have an influence on the lake volume and the water chemistry. Water samples were collected from different points in the lake including spring, brine, and open waters. Samples were collected from these sites in the months of June, July, August, and September 2018. The coordinates of the sampling sites were: S1 (1.891380 S; 36.302632 E), S2 (1.895020 S; 36.299372 E), S3 (1.900988 S; 36.301307 E), S4 (1.908460 S; 36.301996 E), S5 (1.991601 S; 36.258904E), S6 (1.975517 S; 36.236564 E) and BR1 (1.887908 S; 36.300855 E) (Figure 1). S1 was composed of spring water, S2–S6 were composed of open waters, and BR1 was brine. Three sub-samples of 50 ml each were collected from each site and pooled into a composite sample. In addition, water samples for physicochemical analysis were collected. All samples were collected in sterile Conical Centrifuge tubes (Biologix, Shandong, China, Cat. No. 430829) and transported in a cool box (Sp. Berner, Valencia, Spain).
Water temperature, pH, total dissolved solids (TDS), and salinity measurements were recorded in situ. Water temperature, TDS, and salinity were measured using VWR phenomenal handheld Meter (VWR, Atlanta, GA, USA, Model CO 3100H), while pH was measured using Hanna Combo pH meter (Hanna Instruments, Nusafalau, Romania, Model HI-98128). In this case, about 100 ml of sample water was put in a sterile 400 ml glass beaker (Marienfeld, Germany, Cat. No. BR91236). A pre-calibrated meter was dipped in the sample and the readings were recorded. Water samples for dissolved P, K+, NO3-, NH4-, Mg2+, Na-, Fe2-, Ca2+, SO42-, Cl-, and HCO3- measurements were collected in sterile 500ml bottles and stored in a cool box for transportation to Crop Nutrition Laboratory Services (CNLS), Nairobi where analysis was done. Cations such as Ca, Mg, K, Na, Mn, Fe, Cu, Mo, B, Zn, and S were analyzed using atomic absorption spectrometry (AAS), while anion analysis was carried out using mass spectrometry.
Cell biomass for DNA extraction was obtained by centrifuging 50 ml of each water sample at 14,000 rpm for 20 minutes in an Eppendorf centrifuge (Eppendorf, Model 5415R, Cat. Z605212). The pellets were resuspended in 200 μl of a resuspension buffer (25% w/v sucrose (Sigma-Aldrich, Cat. No. S9378) in 50 mM Tris pH 8.5 (Sigma-Aldrich, Cat. No. 93352), and 50 mM EDTA; pH 8.0 (Sigma-Aldrich, Cat. No. 798681). To disrupt the cell wall of Gram positives, 2 μl of lysozyme (20 mg/ml) (Roche, Cat. No. 10837059001) and 10 μl of RNAse A (20 mg/ml) (Roche, Cat. No. 10109142001) were added and incubated at 37°C for 30 minutes. Cell lysis was achieved by the addition of 600 μl of a lysis buffer (1% SDS (Sigma-Aldrich, Cat. No. 8.17034) in 10 mM Tris pH 8.5 (Sigma-Aldrich) and 5 mM EDTA; pH 8.0 (Sigma-Aldrich). The samples were gently mixed with 10 μl of Proteinase K (20 mg/ml) (Sigma-Aldrich, Cat. No. 39450-01-6) and incubated at 65°C for 2 hours. DNA was recovered by adding of an equal volume of chloroform (Sigma-Aldrich, Cat. No. C2432) followed by centrifugation at 13,200 rpm for 10 min at 4°C in an Eppendorf 5415R centrifuge. The aqueous layer was transferred into a new tube 150 μl of sodium acetate (pH 5.2) (Sigma-Aldrich, Cat. No. S8750) and equal volume of isopropyl alcohol (Sigma-Aldrich, Cat. No. 67-63-0). The contents were centrifuged at 13,200 rpm for 10 minutes and the DNA pellet was recovered by washing with 70% ethanol, air dried for 15 minutes and dissolved in 30 μl of nuclease free water (Sigma-Aldrich, Cat. No. 7732-18-5). DNA quality was checked by running an aliquot of 2 μl in 1% agarose (Sigma-Aldrich, Cat. No. A9918) gel electrophoresis (Orwa et al., 2020).
The V4 hypervariable region of the 16S rRNA genes was amplified using the primer 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) (Caporaso et al., 2012). Amplification was done using HotStarTaq Plus Master Mix Kit (Qiagen, USA) under the following cycling conditions: initial denaturation at 94°C for 3 minutes, followed by 35 cycles of denaturation at 94°C for 30 seconds, annealing at 53°C for 40 seconds and elongation at 72°C for 1 minute, after which a final elongation step at 72°C for 5 minutes was performed. Three independent PCR reactions were performed per sample and pooled in equimolar amounts. The PCR products were then checked in a 2% agarose gel. The sample was purified using calibrated Ampure XP beads (Beckman Coulter, Inc., IN, USA). DNA libraries were prepared using Illumina TruSeq DNA libraries (Illumina, Inc., San Diego, CA, United States) and sequencing was performed at MR DNA (Shallowater, TX, USA) on a MiSeq platform (2 × 300 bp) following the guidelines of the manufacturer (Illumina Inc.).
The Q25 sequence data derived from MiSeq sequencing was processed using the MR DNA ribosomal and functional gene analysis pipeline (MR DNA, Shallowater, TX). Sequences were depleted of primers, reads <250 bp and ambiguous base calls were removed. The reads were quality filtered using a maximum expected error threshold of 1.0. Sequences were further processed using VSEARCH v2.14 (Rognes et al., 2016). This included sorting and size-filtering of the paired reads to ≥300 bp (--sortbylength --minseqlength 300) and dereplication (--derep_fulllength). The sequences were then denoised and evaluated for potential chimeric sequences using UCHIME package v.11. (Edgar et al., 2011). Representative operational taxonomic units (OTUs) were picked de novo using VSEARCH v2.14 (Rognes et al., 2016), and assigned taxonomy using BLAST searches against the SILVA v132 rRNA reference database (Quast et al., 2012). A sequence identity cutoff of 97% was used to pick OTUs from the quality-filtered, denoised, non-chimeric sequences. Eukaryotic sequences were filtered from the dataset using the script filter_otu_table.py. in QIIME v1.90 (Caporaso et al., 2010b).
The Illumina raw reads for the 16S rRNA gene sequences were deposited in the Sequence Read Archive (SRA) of NCBI under the accession numbers PRJNA962270 (Kipnyargis et al., 2023b).
Sequences with assigned taxonomy were aligned using PyNast (Caporaso et al., 2010a), and a phylogenetic tree was constructed using FastTree v2.1.7 (Price et al., 2010). The alpha diversity indices (Chao1, abundance-based coverage estimator (ACE), Simpson, Shannon, Fisher’s alpha, Pielou’s evenness, and Good’s coverage) were calculated with QIIME v1.90 (Caporaso et al., 2010) using alpha_rarefaction.py employing the same level of surveying effort (37,000 per sample based on the lowest sample count). All subsequent steps were analyzed in R software v4.2.0 (R Core Team, 2020) and RStudio v1.1.456 (RStudio Team, 2020). The results of all statistical tests were regarded as significant if p 0.05. To compare the (dis) similarity of OTU compositions between communities the OTU abundance table was standardized using decostand (method = “hellinger”). Hierarchical cluster analysis was performed using hclust in R software v4.2.0 (R Core Team, 2020) (method = “average”). The heatmap was created using JColorGrid v1.86 (Joachimiak et al., 2006).
The OTU network generated in QIIME was filtered using an edge cut-off of 0.001 and visualized in Cytoscape v3.9.1 (Otasek et al., 2019) in an “edge-weighted spring-embedded layout”. In this case, sampling sites were used as source nodes and bacterial families as target nodes. Redundancy analysis (RDA), based on Bray dissimilarity was used to test the correlation between the physicochemical parameters and the microbial community at the genus level. This was done using the Microeco package v0.15.0 (Liu et al., 2021) and plotted using the package Pheatmap in R.
To assess the beta diversity of microbial communities, a non-metric multidimensional scaling (NMDS) was performed using Bray-Curtis dissimilarities with the script compare_categories.py. test and weighted UniFrac distance matrix (Lozupone & Knight, 2005) as input using the Vegan package in R (Bray & Curtis, 1957; Oksanen, 2015).
One of the objectives of this study was to investigate the change in water chemistry over time. It has been established that physicochemical factors play a critical role in shaping the structural composition of microbial communities in an ecosystem. Samples from site S1 (spring water) exhibited lower concentrations of the various ions and cations as compared to the other samples. The water temperature ranged from 27°C to 38.7°C (average 33.7°C). The pH of the water was alkaline, ranging from 9.8 (S6_June) to 11.5 (BR1_June) recording the highest pH value of 11.5. The major water cations were Na+ (10,300–160,000 ppm) and K+ (131–4,280 ppm), and the major anions were HCO3− (15,400–277,000ppm) and Cl− (4,050–102,000 mg/L). Phosphorus levels ranged from 2.38–108 ppm, while magnesium and calcium levels were low, ranging from 0.02–16.1 and 0.05–127 ppm, respectively. The total dissolved solids (TDS) ranged from 27.1–153.5 ppm (Table 1).
TDS total dissolved solids, SAR sodium absorption ratio. The samples are denoted as S1 to S6, while BR1 represents the brine sample.
After quality filtering, denoising, and removal of potential chimeras and non-bacterial sequences, approximately 3,197,447 high-quality sequences with an average read length of 525 bp were obtained from the entire dataset. The number of sequences per sample varied from 37,406 (sample S5_Jun) to 285,085 (sample BR1_Sep) with an average value of 121,603 sequences. The number of OTUs per sample varied from 852 (sample S3_July) to 2,024 (sample S5_Sep) (Table 2). All sequences were assigned taxonomy up to genus level and clustered into 4,837 OTUs (97% identity) distributed in the domain Bacteria (3,802 OTUs) and Archaea (1,035 OTUs). Overall, most OTUs were found in S5, while S4 had the least OTUs. The distribution of shared OTUs based on the month of sampling is shown in Extended data, Supplementary Figure 1 (Kipnyargis et al., 2023a).
OTUs, operational taxonomic units; ACE, abundance-based coverage estimator.
The values of the good’s coverage estimator ranged from 81% (S5_Sep) and 96% (S3_Aug) suggesting that the sequencing process captured a significant number of dominant communities. Within the open water samples (S2–S6), site 5 samples collected across the seasons had the highest alpha diversity indices suggesting that S5 had the highest species richness and diversity. S3_Aug samples (open waters) had the lowest alpha diversity indices. Within the hot spring samples (S1), S1 samples collected in September had the highest species richness and diversity. Within the brine samples (BR1), Br1 samples collected in September had the highest species diversity and richness (Table 2).
The alpha diversity indices showed that high microbial diversity was recorded in the month of August, followed by September, June, and July in that order (Figure 2).
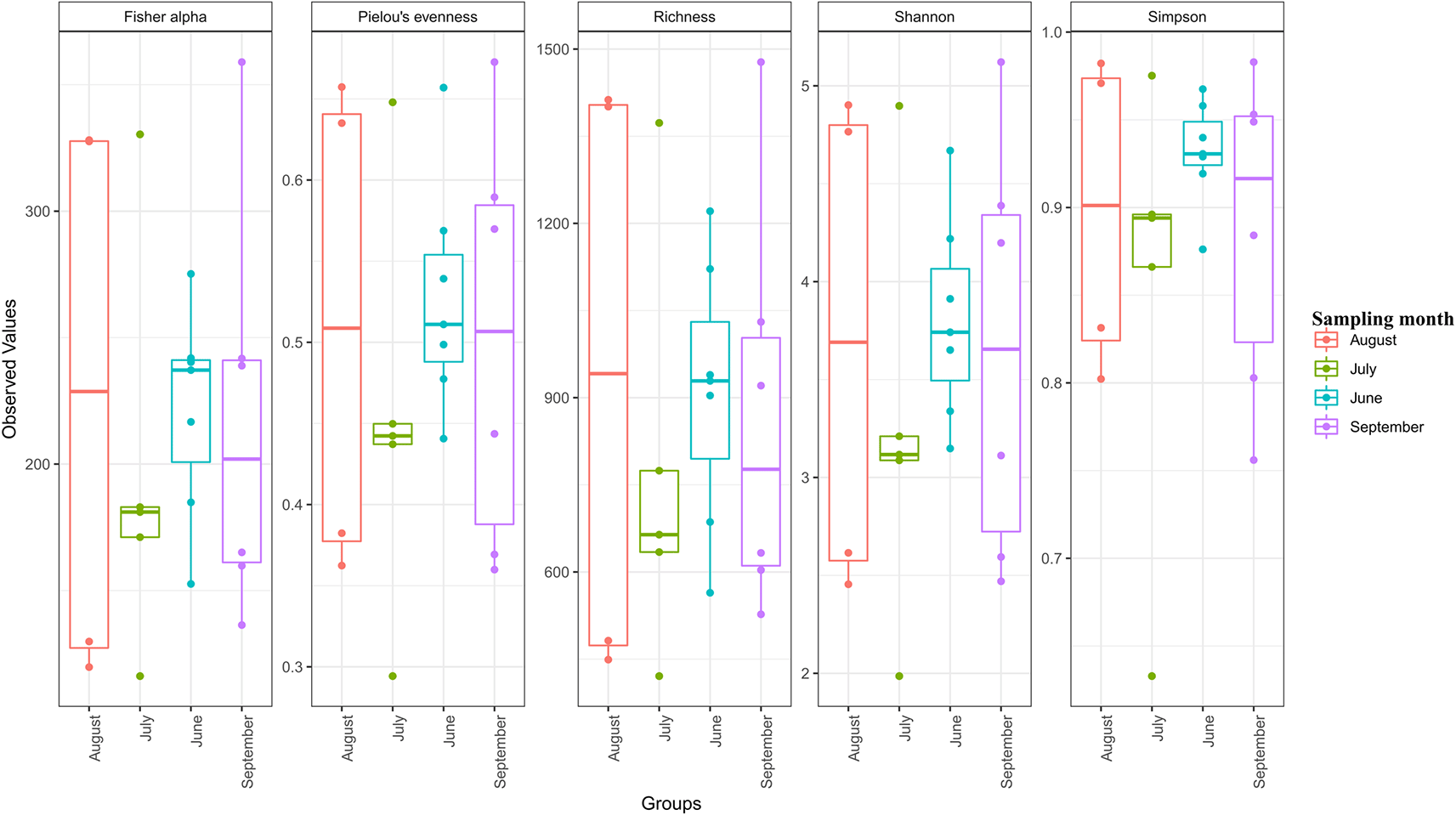
Statistical significance was determined at p < 0.05. Individual sample values and outliers are shown in the form of dots.
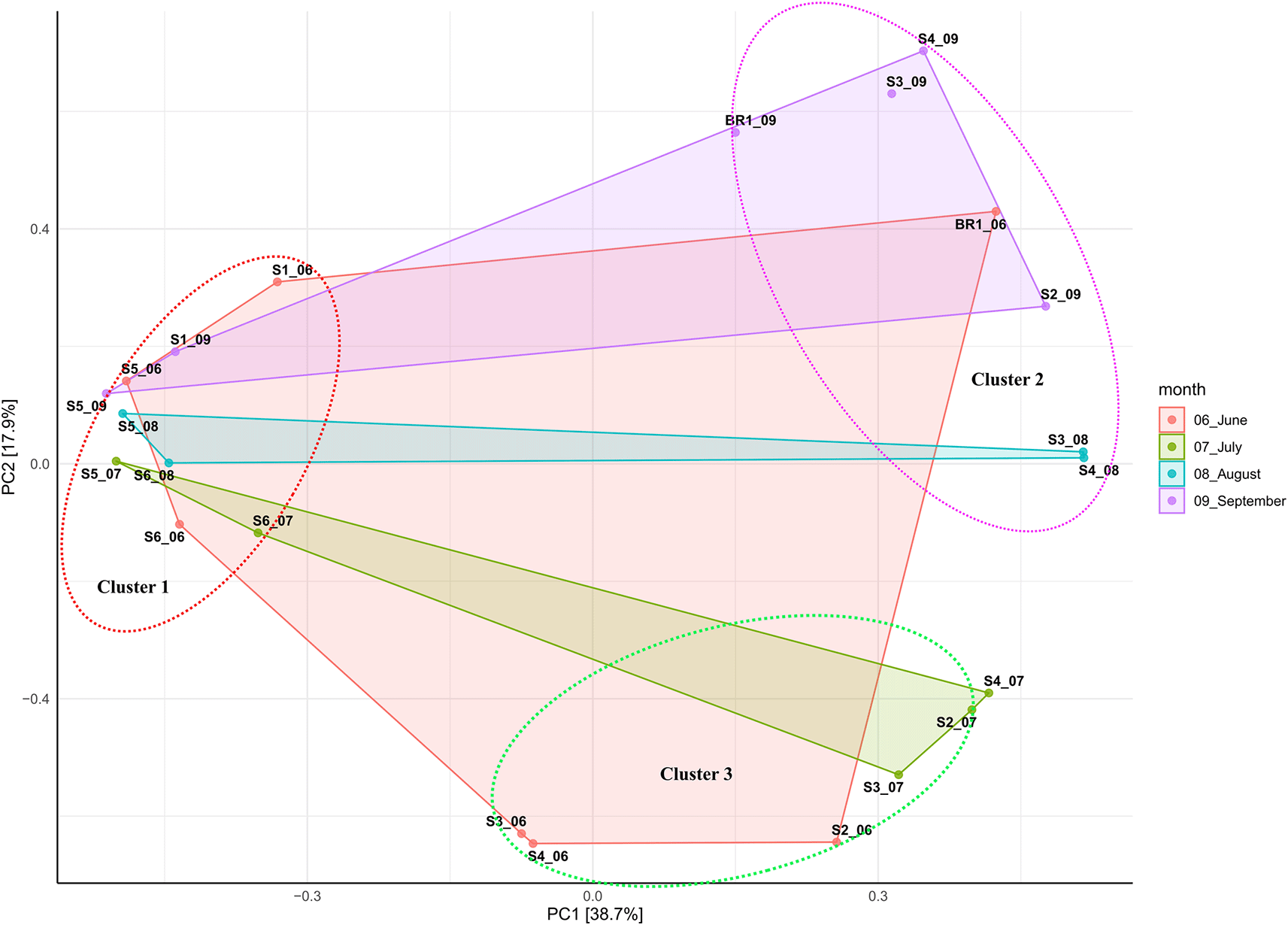
Beta diversity ordination based on Bray-Curtis dissimilarity showed that samples (except hot spring and brine samples) did not cluster based on sampling site. This suggests that hot spring and brine samples better individual community similarity. Overall, all samples clustered together based on salinity and alkalinity, indicating the impact of these elements on the structure of the bacterial and archaeal communities (Figure 3; Table 1). The principal component (PCA) analysis showed that the first (PC1) and second (PC2) axes described 38.7% and 17.9% of the variance in microbial communities, respectively. Accordingly, samples were clustered into three distinct groups based on alkalinity and salinity. Low alkalinity and salinity samples (pH 9.8 – 10.5; 10, 300 ppm – 70,500 ppm) consisted of nine samples (S1_06, S1_09, S5_06, S5_09, S5_08, S5_07, S6_08, S6_06, and S6_07). Moderately alkaline and saline samples (pH 10.5 – 10.6; 63, 900 ppm – 100,000 ppm) consisted of six samples (S3_06, S4_06, S2_06, S3_07, S2_07, and S4_07). Highly alkaline and saline samples (pH 10.7-11.5; >100,000 ppm) consisted of six samples (Br1_06, Br1_09, S4_09, S2_09, S3_08, and S4_08).
The proportion of bacteria to archaea varied by season and sampling month (Figure 4). The results indicate that the archaeal population increased due to evaporative ion concentration while bacteria abundance was higher where the ion concentration was lower (sites 1, 4, and 5) (Table 1). In hot spring water (S1), archaea abundance was the lowest while bacterial abundance was the highest. Within open water samples (S2–S6), S4 had the highest abundance of archaea, while S5 had the highest proportion of bacteria. Within the brine (BR1), the archaea proportion was relatively higher than the bacterial communities. From June to September 2018, bacterial abundance decreased while archaeal abundance increased (Figure 4A).
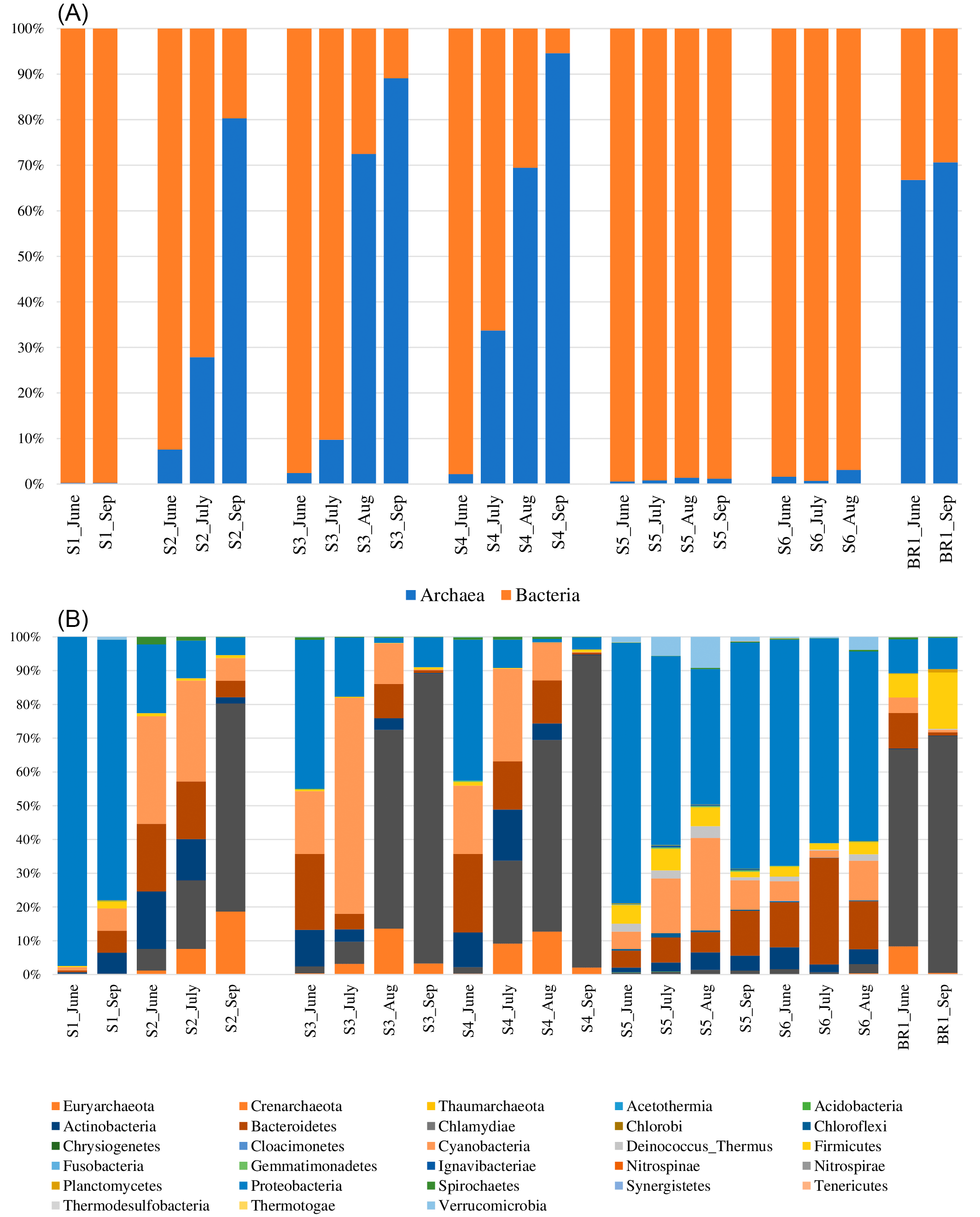
(A) The proportion of Domains bacteria and archaea across the sampling sites and months. (B) Percentage abundance of the most popular bacterial and archaeal phyla across the sampling sites and the sampling months.
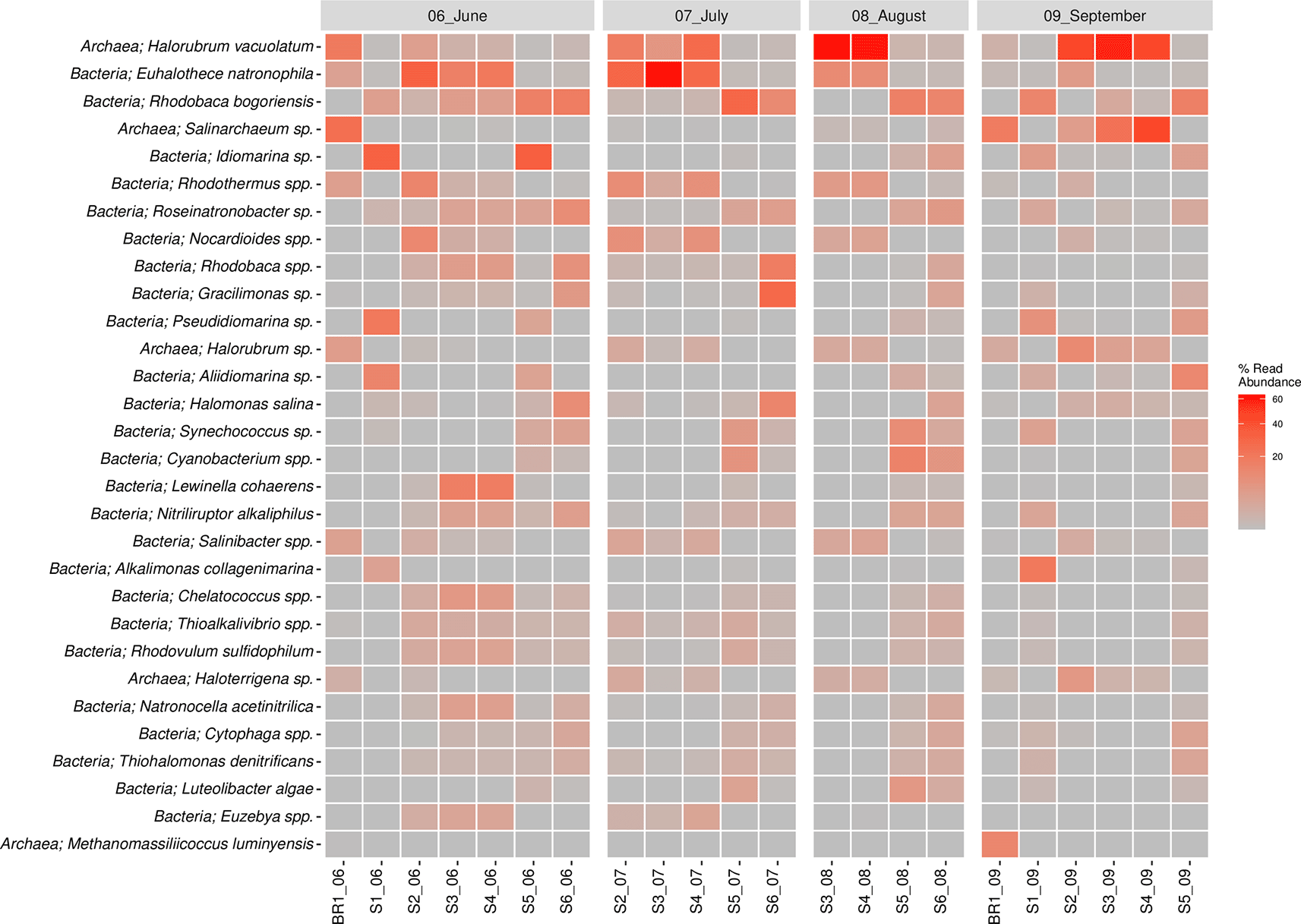
The bacterial reads were distributed across 25 phyla, 107 orders, 225 families, and 545 genera. The results revealed that the most abundant bacterial phyla across the sampling sites and the four months of sampling included Proteobacteria, Cyanobacteria, Bacteroidetes, Actinobacteria, Firmicutes, Verrumicrobia, Deinococcus-Thermus, Spirochaetes, and Chloroflexi (Figure 4B). Notably, members of the phylum Proteobacteria were the most dominant group across all the sampling sites, accounting for 35% abundance. They were followed by phylum Cyanobacteria (14.2%) and Bacteroidetes (10.5%), Actinobacteria (5.2%), Firmicutes (2.7%), and Verrumicrobia (1.1%). The most dominant bacterial genera (> 1% of all sequences across all samples) were Euhalothece (10.3%), Rhodobaca (9.6%), Idiomarina (5.8%), Rhodothermus (3.0%), Roseinatronobacter (2.4%), Nocardioides (2.3%), Gracilimonas (2.2%), Halomonas (2.0%), Lewinella (1.9%), Synechococcus (1.8%), Aliidiomarina (1.8%), Nitriliruptor (1.7%), Thioalkalivibrio (1.7%), Salinibacter (1.4%), Alkalimonas (1.25%), Chelatococcus (1.4%), and Rhodovulum (1.4%). Others included: Cytophaga (0.9%), Natronocella (0.9%), Thiohalomonas (0.9%), Euzebya (0.8%), Paracoccus (0.8%), and Luteolibacter (0.8%). The abundance of bacterial genera was higher in the sampling site S5 (25.3%) followed by S6 with 20.1%, S3 (14.5%), S1 and S4 with 12.8% each, and brine sample BR1 with 3.9% abundance in that order. In terms of the sampling month, June had the highest bacterial abundance with 39.5% followed by the months of July (27%), September (16.8%), and August (16.2%) in that order.
The archaeal reads were affiliated to three phyla (Euryachaeota, Crenarchaeota, and Thaumarchaeota) (Figure 4B), 14 orders, 20 families, and 62 genera. The dominant Phylum was Euryachaeota (87% of all Archaeal samples), with its dominant genera (>1% of all sequences across all samples) being Halorubrum (18.3%), Salinarchaeum (5.4%) and Haloterrigena (1.3%). Other genera included Methanomassiliicoccus (0.6%), Palaeococcus (0.4%), Halovenus (0.3%), Thermococcus (0.3%), Haladaptatus (0.3%), Halorientalis (0.3%), Methanobrevibacter (0.2%), Natronomonas (0.2%), Halohasta (0.2%), Haloquadratum (0.1%), and Methanobacterium (0.1%). Archaeal genera abundance was higher in the sampling site S3 (27.2%) followed by brine site BR1 with 21.6% abundance. S1 and S6 had the least archaeal abundance with 0.08 and 0.8%, respectively. In terms of the sampling month, September had the highest archaeal abundance with 53% followed by August (23%), June (12.7%), and July (11.4%).
The bacterial species composition (>1%) included Euhalothece spp. (10.3%), Rhodobaca spp. (9.6%), Idiomarina spp. (5.8%), Rhodothermus spp. (3.0%), Roseinatronobacter spp. (2.4%), Nocardioides spp. (2.3%), Gracilimonas spp. (2.2%), Halomonas sp. (2%), Lewinella (1.9%), Synechococcus spp. (1.8%), Cyanobacterium spp. (1.8%), Aliidiomarina spp. (1.7%), Nitriliruptor spp. (1,7%), Thioalkalivibrio spp. (1.7%), Salinibacter spp. (1.4%), Alkalimonas spp. (1.2%), Chelatococcus spp. (1.1%), and Rhodovulum spp. (1.1%). The Euhalothece natrophila species were abundant in June, July, and August, except in sites S5 and S6 across all seasons. Rhodobaca bogoriensis was largely sampled in the month of June and site S6 in July and August 2018. Idiomarina spp. were largely concentrated in the months of June, particularly in sites S1 and S5, whereas Rhodovulum spp. were sampled across all seasons. Lewinella coherens were sampled in June mostly in sites S3 and S4. On the other hand, Halorubrum spp. (18.3%), Salinarchaeum spp. (5.3%), Haloterrigena spp. (1.3%), Methanomassiliicoccus spp. (0.7%), and Palaeococcus spp. (0.5%) were the major species in the Archaeal Domain. Idiomarina vacuolatum was sampled across all the sampling seasons but varied in structure across the sampling sites. Halorubrum vacuolatum was mainly sampled in the month of August (S1 and S2) and September (S3, S4 and S5). Salinarchaeum sp. was mainly sampled in the month of September, while Haloterrigena spp. was sampled across the seasons and sites, though in low proportions. The top 30 most abundant species of bacteria and archaea are shown in Figure 5. Overall, Halorubrum spp. was the most abundant species sampled followed by Euhalothece spp. and Rhodobaca spp. (Extended data, Supplementary Figure 2 (Kipnyargis et al., 2023a)).
Microbial co-occurrence network analysis at the family level revealed that bacterial members of the family Cyclobacteriaceae, Burkholderiaceae and Alteromonadaceae were unique to the S1 sampling site. Bacterial members of Halobacteroidaceae, Spirochaetaceae, halanaerobiaceae, and desulfohalobiaceae, as well as Archaeal members of Archaeoglobaceae and Methanobacteriaceae, were found exclusively in the S2 sampling site. Phyllobacteriaceae and Nostocaceae were unique to S3, while Alcanivoracaceae, Carnobacteriaceae, and Marinilabiliaceae bacteria were found in S5 only. Unique to S6 were the bacterial Puniceicoccaceae family. The highest number of co-shared families was found between S5 and S1 co-sharing eight families, whereas Bacilaceae and Natranaerobiaceae were found in S5 and S2, and S6 and S2 co-shared only one family (Pseudomonadaceae) (Figure 6).
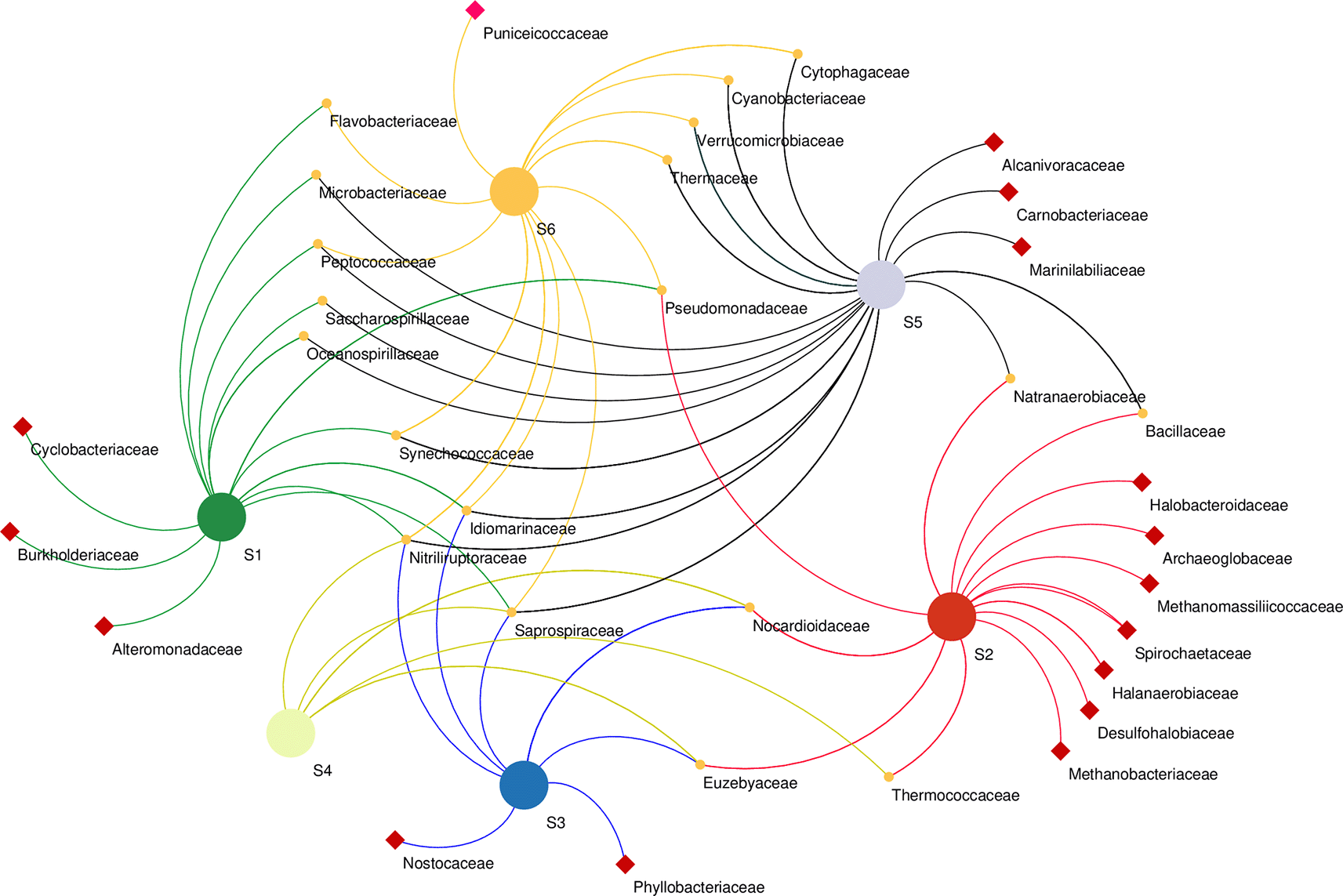
Samples marked with red squares indicate their exclusive site of isolation.
Redundancy analysis (RDA) was used to assess the effect of water chemistry on microbial community structure in Lake Magadi. The results reveal that changes in the physicochemical parameters influenced the microbial communities in the lake (Extended data, Supplementary Table 2 (Kipnyargis et al., 2023a)). The RDA explained 62.2% and 17.2% of the variation in the first (RDA1) and the second (RDA2) axes, respectively (Figure 7).
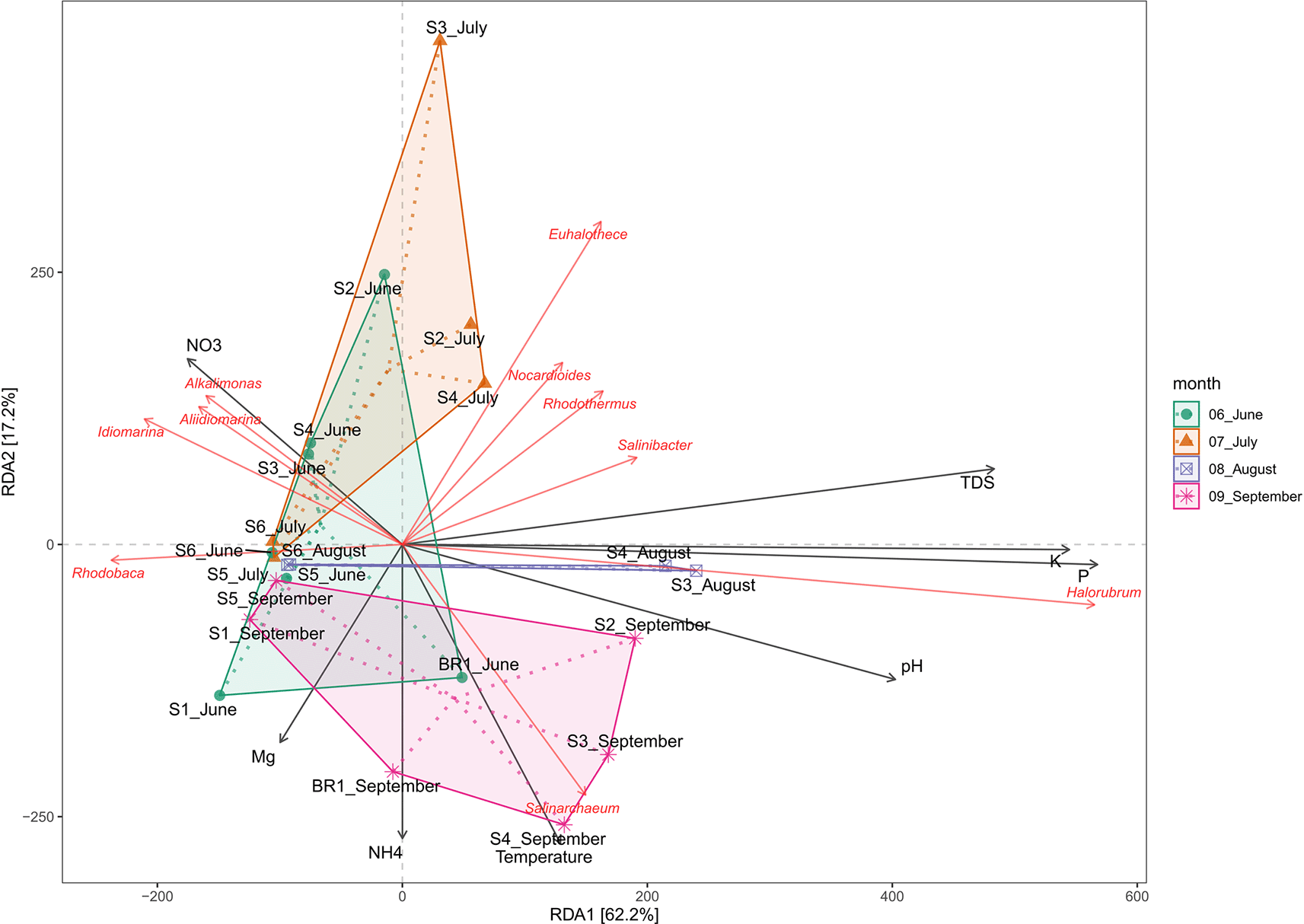
Overall, temperature, pH, P+, K+, NO3-, and TDS had a positive influence on the microbial community structure. Generally, members of genera Nocardiodes, Rhodothermus, Haloterrigena, Methanomasiliicoccus, Halorubrum, Palaeococcus, Nocardioides, Salinarcheum, Salinibacter, and Euhalothece spp. had a wide range of adaptability. Conversely, Synechococcus, Thioalkalivibrio, Cyanobacterium, Rhodovulum, Lewinella, Idiomarina, Pseudidiomarina, Chelatococcus, Aliidiomarina, and Alkalimonas spp. were adapted to fewer physicochemical factors (Extended data, Supplementary Figure 3 (Kipnyargis et al., 2023a)). Notably, Halorubrum and Haloterrigena spp. were positively correlated with P and K (R2 = 0.66, p < 0.001), but negatively correlated with Mn+ and CO32-. pH appears to positively correlate with the structure of the members of the genus Salinarcheum (R2 = 0.245; p < 0.004), but negatively correlated with NO3-. Members of Alkalimonas, Idiomarina, and Aliidiomarina spp. were positively correlated with NO3- (R2 = 0.049, p < 0.210), but negatively correlated with all other tested parameters. Members of Nocardiodes, Rhodothermus, Salinarcheum, Salinibacter, and Euhalothece spp. were positively correlated with total dissolved solids (TDS), alkalinity, salinity, CO32+, and NH4+ (R2 = 0.606, p < 0.001), but negatively correlated with Mg2+, Mn+, and NO3-. On the other hand, Mn+, temperature CO32-, and NH4+ negatively affect the structure of Rhodobaca.
We explored the structure and composition of microbial communities based on the seasonality and physicochemical parameters of Lake Magadi. The physicochemical parameters revealed high concentrations of sodium salts, HCO3-, SO42-, pH values of 9.8–11.5, temperatures of 27–38°C, and low concentrations of Ca2+, Mg2+, and Cu2+. These findings were consistent with previous reports indicating that soda lakes are characterized by moderate to high temperatures, high concentrations of HCO3−/CO32− and reduced concentrations of Ca2+ and Mg2+ (Sorokin et al., 2014a; Vavourakis et al., 2018). However, total dissolved solids (TDS) ranged from 27 ppm (0.02g/L) to 143 ppm (0.143g/L), a situation that is lower than other soda lakes (Taher, 1999; Hosam et al., 2017; Pérez & Chebude, 2017). Furthermore, sulfate concentration (39–958 ppm) was lower than that of lakes Sidi Ameur and Himalatt (Algeria) (Boutaiba et al., 2012). The concentrations of these elements (except pH) were varied across the sampling months and sites, suggesting that the lake chemistry is constantly changing in its constituent elements. Hypersaline lakes are characterized by high amounts of Na2CO3 and NaHCO3 that maintains constant pH in these ecosystems (Simachew et al., 2016). It is hypothesized that Ca2+ and Mg2+ precipitate as insoluble carbonates due to high evaporation rates in these ecosystems. As a result, an alkaline brine with Na+, Cl−, and HCO3−/CO32− accumulates as main ions. The shift in CO2/HCO3−/CO32− equilibrium towards CO32−, leads to the formation of a soda (Na2CO3) lake with pH values of over 10.0 (Grant and Jones, 2016).
Under the extremes of salinity and alkalinity, microorganisms in soda lakes have devised mechanisms of coping up with osmotic stress. For instance, bacteria possess Na+/H+ and K+/H+ antiporters which exchange cytoplasmic cations for protons outside of the cell to achieve lower cytoplasmic alkalinity than the external environment (Padan et al., 2005) Furthermore, they synthesize compatible solutes like betaine glycine, sugars (such as trehalose), polyols, amino acids, biosurfactants, and ectoines, which are involved in maintaining an isotonic environment (Bursy et al., 2008). The archaea produce osmolytes such as include glycerol, glycosyl glycerol, betaines, proline, glutamate, glutamine, and ectones (Roberts, 2005).
A high diversity of OTUs was detected for the domain Bacteria with 3,802 OTUs while Archaea had 1,035 OTUs. Bacterial diversity was dominated by the phyla Proteobacteria, Cyanobacteria, Bacteroidetes, Actinobacteria, Firmicutes, Verrumicrobia, Deinococcus-Thermus, Spirochaetes, and Chloroflexi. Similar results have been shown from soda ecosystems such as Solar saltern in Tunisia (Menasria et al., 2019), lake Chott El Jerid (Abdallah et al., 2018), hot springs of Lake Magadi (Kambura et al., 2016), and lakes Sonachi, Magadi, Elmenteita, and Bogoria in Kenya (Mwirichia, 2022). Notably, the phylum Proteobacteria was the most dominant group across all the sampling sites, accounting for 35% abundance. The function of members of Proteobacteria such as Burkholderiaceae is to decompose recalcitrant organic matter while others like Beijerinckiaceae fix atmospheric nitrogen (Li et al., 2012). They were followed by the phylum Cyanobacteria (14.2%) represented mainly by the Euhalothece spp. Euhalothece is a single-celled stenohaline cyanobacterium that grow optimally at 7% (w/v) NaCl. They depict a morphological variability depending on the concentrations of NaCl and carbonates as well as the pH conditions (Mikhodyuk et al., 2008). In soda lake ecosystems, Arthrospira spp. are the main photosynthetic agents driving primary productivity, although the seasonal occurrence of Cyanospira, Synechococcus, and Chroococcus spp. augment this process (Jones et al., 1998). In soda lakes, Cyanobacteria are the major contributors to nitrogen fixation (Sorokin & Kuenen, 2005). Phylum Bacteroidetes was majorly represented by the genera Rhodothermus, Roseinatrobacter, Gracilimonas, Lewinella, and Cytophaga. Verrumicrobia was represented by Verrucomicrobium, Puniceicoccus, and Coraliomargarita. Members of Bacteroidetes and Verrucomicrobia thrive well in high-nutrient environments where they play a role in the degradation of biopolymers such as cellulose and chitin (Newton et al., 2011). Interestingly, the presence of Bacteroidetes was often associated with the availability of Cyanobacteria across the sampling periods (Extended data, Supplementary Table 1 (Kipnyargis et al., 2023a)). Phylum Firmicutes was represented by members of Class Clostridium (Clostridium, Halanaerobium, Natranaerobius, and Moorella sp.) and Bacilli. (Alkalibacterium and Anoxybacillus sp.). The two Classes are common in soda lakes with the addition of Tenericutes in some instances.
Our findings showed a shift in bacterial composition throughout the sampling seasons where a high abundance of Actinobacteria and Proteobacteria was accompanied by a lower abundance of Actinobacteria. A typical feature of Actinobacteria is their sensitivity to nutrient overloading and subsequent reduction in oxygen levels. On the other hand, Proteobacteria, Firmicutes, and Bacteroidetes are adapted to nutrient overloading and degradation of complex biopolymers and dead organic materials (DOM) (Thomas et al., 2011). However, the abundance of Firmicutes, which possess diverse metabolic capacities and are resistant to oxygen limitations (Martiny et al., 2006) depicted an uneven behavior in relation to Actinobacterial composition.
The archaeal community diversity in the Lake Magadi microbiome was represented by three phyla, the Euryachaeota, Crenarchaeota, and Thaumarchaeota. Generally, archaea were more in brine samples with Br1_June and Br1_Sept accounting for 24.5% of total archaea. Previous studies have indicated that archaea are more adapted to saline environments than bacteria (Mani et al., 2020). Euryachaeota was the most abundant phylum across the sites and the sampling seasons. The phylum Euryachaeota accounted for 87% of all archaeal communities. Euryachaeota has well-adapted inhabitants of hypersaline environments where they play a critical role in ecosystem services such as carbon cycling by functioning as methanogens (Jiang et al., 2007; Vavourakis et al., 2016). Grant et al. (1999) first characterized this phylum the alkaline saltern of Lake Magadi. The second most abundant group was represented by Thaumarchaeota also known as ammonia-oxidizing agents (Andreote et al., 2018). Their distribution along a salinity gradient in estuarine sediments may be linked to changes in location and/or salinity as well as gradient sediment depth (Webster et al., 2015). These results were consistent with those of other soda lakes that have detected members of the phylum Euryachaeota and Crenarchaeota (Ghori et al., 2021), Crenarchaeota, Euryarchaeota, Woesearchaeota, and Pacearchaeota, Euryachaeota and Woesearchaeota (Wang et al., 2022). The most abundant genera belonged to Halorubrum (18.3%), Salinarchaeum (5.4%), and Haloterrigena (1.3%). Most of these microbes have their habitats in soda lakes and neutral saline environments (Feng et al., 2005; Mwatha & Grant, 2016; Minegishi et al., 2017; Zhao et al., 2020). Other haloalkaliphilic archaea related to genera Natronomonas, Natrialba, Natrococcus, Natronobacterium, Natronolimnobius, and Halorubrum all of whom were detected in this study, have previously been isolated from brines of East African soda lakes and Inner Mongolian lakes where salinity values reach > 30%, and pH values of >10 (Grant and Sorokin, 2011). Overall, the results of this study reflect bacterial composition in many soda lakes around the world (Sorokin et al., 2014a; Kambura et al., 2016; Mani et al., 2020; Poyraz & Mutlu, 2020; Wang et al., 2022).
Co-occurrence network analysis demonstrates the interactions between microbial taxa, which can be symbiotic or competitive (He et al., 2019). At the family level revealed the presence of heterogenous microbial communities that co-occur in different sampling sites along the lake as well as others that were unique to a particular site, suggesting an ecological adaptation of these communities to certain aspects of their sites. For instance, Desulfobacteriaceae were unique to S2. Correspondingly, S2 had the highest concentration of both sulfur and sulfate ions (Table 1). This family, particularly Desulfonatronum, Desulfonatronospira, Desulfonatronovibrio, and Desulfohalophilus have been shown to thrive in anoxic parts of soda lakes acting as sulfate-reducing bacteria (SRB) through the oxidation of hydrogen and formate or direct disproportionation of sulfite of thiosulfate (Sorokin et al., 2011). Remarkably, Thioalkalivibrio sp. was not significantly affected by the physicochemical properties investigated. This group of sulfur-oxidizing bacteria (SOB) has been suggested to have a wide range of adaptation mechanisms in soda lake ecosystems (Li et al., 2022). Unique to the S6 site were the members of the family Puniceicoccaceae which have also been described in four soda lakes of the Cariboo Plateau in Canada (Zorz et al., 2019). Cyclobacteriaceae retrieved from the S1 site have established habitats in diverse ecosystems like cold marine regions like algal/microbial mats, haloalkaline soda lakes, Antarctica, freshwater bodies, marine waters, marine sediments, mangroves, hot springs, and mud volcanoes (Rosenberg et al., 2014). Members of families Rhodobacteraceae and Cyclobacteriaceae have sulfate–oxidizing properties, whereas Burkholderiaceae (unique to S1) have adapted to different ecological niches and are involved in processes such as catabolism of aromatic compounds as well as nitrogen fixation (Pérez-Pantoja et al., 2012).
Alpha diversity studies revealed that samples in the open waters, particularly S5, had the highest species richness and diversity. However, open waters samples from S2–S4 depicted varying degrees of community structure and this could be due to variations in the intertidal water zones (Zhu et al., 2018). Brine samples (BR1) in June and September had relatively high diversity and evenness indices. This phenomenon can be attributed to the maintenance of homogenous abundances by microbial communities during brine formation, hence resulting in higher biodiversity (Banda et al., 2020). Hot spring samples collected in September showed high species diversity indices. Microbial samples from Lake Magadi hot springs were established to be stable and active (Kambura et al., 2016).
Beta diversity analysis based on principal component analysis (Figure 3) revealed that samples from hot spring (S1) and brine (BR1) clustered according to their sites. This suggests that hot spring and brine samples had better individual community similarity (Tao et al., 2019). However, the salinity and alkalinity of the sampling sites appeared to drive the overall dynamics of microbial community structure in Lake Magadi. Previous studies have established that salinity is the primary selective force driving the distribution of beta diversity, whereas alkalinity drives microbial richness (Antony et al., 2013; Boros & Kolpakova, 2018; Banda et al., 2020). Moreover, extreme salinity and alkalinity confine the microbial communities to a few taxa highly adapted to the prevailing conditions (Oren, 2011).
In terms of water chemistry (Figure 7; Extended data, Supplementary Figure 3 (Kipnyargis et al., 2023a)), pH, temperature, PO43-, K+, and NO3−, NH4+, Mn+, Na+, SO42-, and TDS influenced the variation of microbial community composition in Lake Magadi. Salinity and alkalinity tend to influence the richness of the microbial communities in the soda lake ecosystem. Specifically, members of genera Nocardiodes, Rhodothermus, Haloterrigena, Methanomasiliicoccus, Halorubrum, Palaeococcus, Nocardioides, Salinarcheum, Salinibacter, and Euhalothece had a wide range of physicochemical adaptability. Conversely, Synechococcus, Thioalkalivibrio, Cyanobacterium spp., Rhodovulum, Lewinella, Idiomarina, Pseudidiomarina, Chelatococcus, Aliidiomarina, and Alkalimonas adapted fewer physicochemical factors (Extended data, Supplementary Figure 3 (Kipnyargis et al., 2023a)). The archaeal genera Salinarchaeum and Halorubrum Halobellus, Halolamina, Methanobrevibacter, and Halorhabdus have been strongly associated with salinity and other factors such as pH, Mg2+, Na+, K+, Ca2+, and SO42– (Han et al., 2017). Nitrate appears to drive the members of the genera Aliidiomarina, Idiomarina, and Alkalimonas (Figure 4, Extended data Supplementary Figure 3 (Kipnyargis et al., 2023a)). Many strains of Alkalimonas have been isolated from Chahannor (China), Kulunda Steppe (Russia), and Elementaita (Kenya) soda lakes where they play a role in nitrate reduction and formation of H2S (Ma et al., 2004; Vavourakis et al., 2016). However, in this study, sulfur was negatively correlated with Alkalimonas. The Aliidiomarina and Idiomarina belong to family Idiomarinaceae and have also been described as nitrogen reducers but poor in carbohydrate utilization (Chiu et al., 2014).
Studies involving ecological, physiological, and taxonomical aspects have revealed the great diversity of haloalkaliphiles in numerous saline and alkaline lakes. The current study utilized a culture-independent technique to elucidate the microbial community structure and composition based on different sampling periods across various sites of the saline and alkaline Lake Magadi. The results depict a great deal of diversity in bacterial diversity as compared to archaea. Salinity and alkalinity are the main drivers of the microbial community in the lake. Overall, members of Nocardiodes, Rhodothermus, Haloterrigena, Methanomasiliicoccus, Halorubrum, Palaeococcus, Nocardioides, Salinarcheum, Salinibacter, and Euhalothece had a wide range of adaptability. Conversely, Synechococcus, Thioalkalivibrio, Cyanobacterium spp., Rhodovulum, Lewinella, Idiomarina, Pseudidiomarina, Chelatococcus, Aliidiomarina, and Alkalimonas were affected by fewer physicochemical factors. Additionally, we found that other physicochemical parameters such as TDS, temperature, salinity, alkalinity, nitrates, and phosphates have a positive correlation with microbial community structure. Future research should focus on the functional profiles of samples during and after cyanobacterial blooms, including vertical profile stratification of Lake Magadi. The inclusion of sediment samples will also elucidate the taxonomic and functional profile of anoxic microbial communities.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)