Keywords
Genome assembly, reference genome, transcriptome, Aves, mitogenome
This article is included in the Genomics and Genetics gateway.
Genome assembly, reference genome, transcriptome, Aves, mitogenome
We have addressed the reviewers comments around the mitogenome assembly and addressed the other minor comments.
See the authors' detailed response to the review by Phred Benham
See the authors' detailed response to the review by Charles Feigin
The swift parrot (Lathamus discolor) is a migratory parrot that breeds on the eastern seaboard of the island of Tasmania, Australia and winters on southeastern mainland Australia (Kennedy & Tzaros, 2005; MacNally & Horrocks, 2000; Saunders & Heinsohn, 2008). The swift parrot is Critically Endangered (BirdLife International, 2018) due to the combined effects of logging of its important breeding habitat (Webb et al., 2019) and the impacts of an introduced predator, the sugar glider (Petaurus breviceps) (Heinsohn et al., 2015). Population viability analysis has shown that the already small population of only a few hundred swift parrots (Olah et al., 2021) is likely to rapidly decline over coming generations (Heinsohn et al., 2015; Owens et al., 2023) Although the species has already been subject to population genetic study (Olah et al., 2021; Stojanovic et al., 2018), there remain outstanding questions about multiple aspects of the species’ genetic ecology. For example, like other parrots with small population sizes (Morrison et al., 2020), understanding the genetic basis of immune competence is critical for managing demographic impacts of disease in swift parrots (Saunders & Tzaros, 2011). To facilitate detailed genomic research on this species, we sequenced DNA with PacBio long reads to generate a draft reference assembly and sequenced RNA from five tissues to provide transcriptomic resources to assist in genome annotation for the swift parrot.
A single captive bred female swift parrot died as a result of liver infection. Tissue samples were dissected and flash frozen at -80°C or preserved in RNAlater before being frozen at -80°C. High molecular weight (HMW) DNA was then extracted from heart and kidney tissue using the Nanobind Tissue Big DNA Kit v1.0 (Circulomics: SKU 102-302-100) using the standard protocol. A Qubit fluorometer was used to assess the concentration of DNA with the Qubit dsDNA BR assay kit (Thermo Fisher Scientific). Total RNA was extracted from gonad, spleen, liver, heart and kidney using the RNeasy Plus Mini Kit (Qiagen: 74134) with RNAse-free DNAse I set (Qiagen: EN0521) using the standard protocol. RNA quality was determined using the NanoDrop (Thermo Fisher Scientific) and RNA integrity (RIN) score determined using the Bioanalyzer RNA nano 6000 kit (Agilent 2100: 5067-1511).
HMW DNA was sent for Pacific Biosciences High Fidelity (PacBio HiFi) library preparation with the SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences: 101-853-100) and sequencing on one single molecule real-time (SMRT) cell of the PacBio Sequel II at the Australian Genome Research Facility (St Lucia, Australia). Total RNA from the heart, gonad, kidney, liver and spleen was sequenced as 100 bp paired-end (PE) reads using an Illumina Novaseq 6000 with Illumina Stranded mRNA library preparation at the Ramaciotti Centre for Genomics (University of New South Wales, Kensington, Australia).
The genome assembly was conducted on the Galaxy Australia public server usegalaxy.org.au (Afgan et al., 2016) running the Genome assembly with ‘hifiasm’ (RRID:SCR_021069) (Cheng et al., 2022) on Galaxy Australia workflow v2.1 (Price & Farquharson, 2022). Briefly, Picard (http://broad institute.github.io/picard) (Galaxy version 2.18.2.2; RRID:SCR_006525) SamToFastq, samtools (Danecek et al., 2021; Li et al., 2009) (Galaxy version 2.0.3; RRID:SCR_002105) flagstat and fastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) (Galaxy version 0.72; RRID:SCR_014583) was used to convert BAM files to FASTQ and quality check the reads for input to Hifiasm (Cheng et al., 2022). Hifiasm, with default parameters (Galaxy version 2.1), was run on Galaxy Australia to assembly the genome. Basic genome assembly statistics were calculated with the stats.sh script in BBMap (sourceforge.net/projects/bbmap/) (RRID:SCR_016965). Genome completeness was determined using Benchmarking Universal Single-Copy Orthologues (BUSCO; RRID:SCR_015008) v5.4.6 (Simao et al., 2015) with the vertebrata_odb10 (n = 3354) and aves_odb10 (n= 8338) lineage on Galaxy Australia. Repetitive elements of the genome were identified, classified and masked using a Pawsey Supercomputing Centre Nimbus cloud machine (256GB RAM, 64 vCPU, 3 TB storage) by building a database using RepeatModeler v2.0.1 (RRID:SCR_015027) (Flynn et al., 2020); repeats were then masked using RepeatMasker v4.0.9 (RRID:SCR_012954) (Smit et al., 2013-2015) with the -nolow parameter to avoid masking low complexity repeats.
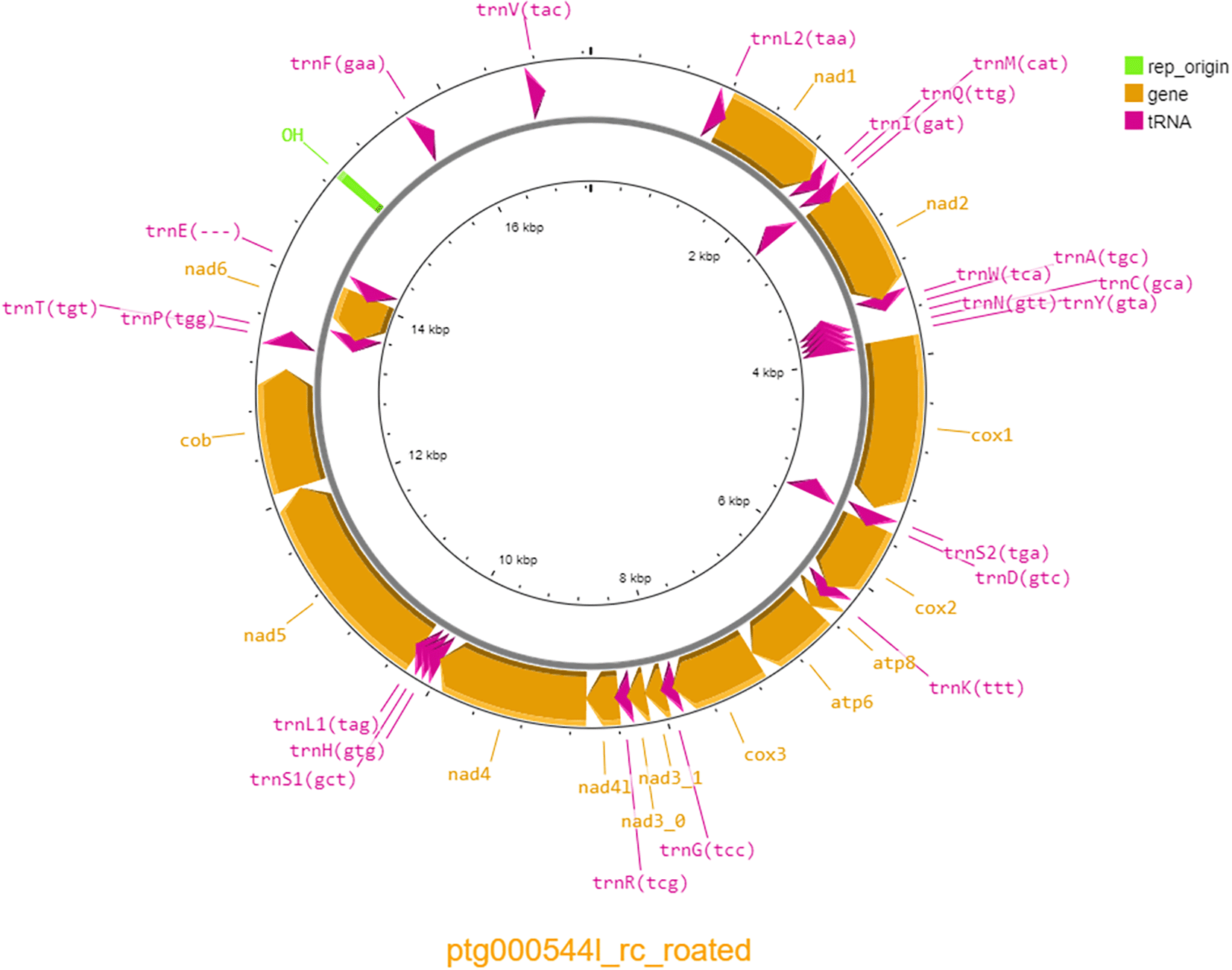
The mitochondrial genome was identified from the reference genome assembly using MitoHiFi v2 (Allio et al., 2020; Uliano-Silva et al., 2023). MitoHifi identified the most taxonomically closely related publicly available mitochondrial genome as the thick-billed parrot (Rhynchopsitta pachyrhyncha) (NCBI reference sequence OR209192.1). The mitochondrial reference sequence for the thick-billed parrot was then used to search for the swift parrot mitochondrial genome. The identified mitochondrial sequence was then added to the genome assembly and annotated using MITOS v 2.1.7 (Donath et al., 2019) and visualised using Proksee (Grant et al., 2023).
Transcriptome assembly was performed on the University of Sydney High Performance Computer, Artemis. Raw transcriptome reads were quality assessed pre and post trimming with FastQC v0.11.8 (RRID:SCR_014583). Trimmomatic v0.39 (RRID:SCR_011848) (Bolger et al., 2014) with the parameters SLIDINGWINDOW:4:5, LEADING:5, TRAILING:5 and MINLEN:25 and ILLUMINACLIP:2:30:10 with the TruSeq3-PE adapters was used to quality trim reads. The repeat masked genome was indexed and trimmed reads aligned using the -dta parameter with hisat2 v2.1.0 (RRID:SCR_015530) (Kim et al., 2019). Resulting sam files were converted to bam format and sorted using samtools v1.9 (Danecek et al., 2021). Stringtie v2.1.6 (RRID:SCR_016323) (Pertea et al., 2015) was used to generate a GTF for each transcriptome. Stringtie v2.1.6 with the -merge parameter merged transcripts into a global transcriptome retaining only transcripts with an FPKM > 0.1 and length > 30. CPC2 v2019-11-19 (Kang et al., 2017) was used to predict coding potential and only transcripts predicted to be coding were retained. Transdecoder v2.0.1 (https://github.com/TransDecoder/TransDecoder) (RRID:SCR_017647) was used to predict open reading frames in the global transcriptome with a minimum transcript length of 20. Transcriptome completeness was assessed using BUSCO v5.4.6 (Simao et al., 2015) with the vertebrata_odb10 (n = 3354) and aves_odb10 (n = 8338) lineage on Galaxy Australia.
Genome annotation was performed using FGENESH++ v7.2.2 (Softberry; RRID:SCR_018928 (Solovyev et al., 2006)) using the longest open reading frame as predicted from the global transcriptome, non-mammalian settings and optimised parameters supplied with the American crow (Corvus brachyrhynchos) gene finding matrix, which is the closest related species with a gene finding matrix provided by FGENESH++. BUSCO v5.4.6 (Simao et al., 2015) in protein mode was run on Galaxy Australia to assess the completeness of the annotation with the vertebrata_odb10 (n = 3354) and aves_odb10 (n = 8338) lineage. The ‘genestats’ script (https://github.com/darencard/GenomeAnnotation) was used to obtain the average number of exons and introns and the average exon and intron length.
Genome assembly using Hifiasm with PacBio HiFi data from a single SMRT cell resulted in a coverage of 28.7x and a genome of 1.24 Gb in size consisting of 847 contigs with a contig N50 of 18.97 Mb and L50 of 20 contigs. The genome assembly was also highly complete with 97.0% of aves_odb10 complete BUSCOs identified (Table 1). The mitochondrial genome was 17,265 bp long and contained 38 genes, including 22 tRNAs and 14 protein coding genes, with a GC percentage of 44.88% (Figure 1).
Trimming retained greater than 99.95% of raw reads which were then aligned to the repeat-masked reference genome. Individual tissue transcriptomes had variable mapping rates from 31.04% for heart tissue to 82.76% for gonad tissue (kidney: 62.26%, liver: 78.84%, spleen: 73.60%). The alignment rate for the heart tissue was low so we excluded heart transcripts from downstream analysis. The poor performance of the heart tissue is potentially due to the comparatively lower concentration of RNA in the heart tissue extraction (35.2 ng/μl) compared to the other 4 tissues (average = 1243 ng/μl [SD: 481]) and the heart tissue was not stored in RNAlater. After using stringtie -merge to generate a global transcriptome and filtering on coding potential and open reading frames with CPC2 and transdecoder, respectively, 14,045 longest open reading frame transcripts were used as mRNA evidence to guide genome annotation. The global transcriptome had 90.8% complete aves_odb10 BUSCOs (Table 2). A total of 27,867 genes were predicted from genome annotation, higher than the predicted 15,000-16,000 genes in birds (Zhang et al., 2014). The annotation contained 78.1% complete aves_odb10 BUSCOs (Table 2). Repetitive elements comprised 17.25% of the genome, mainly consisting of long interspersed elements (LINEs), comparable with other bird genomes (Zhang et al., 2014) (Table 3).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)