Keywords
Aculifera, Aplacophora, Solenogastres, genome
This article is included in the Genomics and Genetics gateway.
This article is included in the Nanopore Analysis gateway.
Aculifera, Aplacophora, Solenogastres, genome
With their incredible diversity in morphology, life history and ecology, molluscs are of great interest to diverse fields of biology ranging from ecology to evolutionary developmental biology to biomechanics. To date there are high-quality genomes available for six of the eight molluscan classes publicly available, although sampling is heavily biased toward the economically and ecologically important members of the clade Conchifera (e.g. Gastropoda, Bivalvia and Cephalopoda; reviewed by Gomes-dos-Santos et al. 2020 and Sigwart et al. 2021). As the sister taxon to all other molluscs, the clade Aculifera (Polyplacophora + Aplacophora) is important to our understanding of molluscan evolution and innovation. Despite their importance, only three aculiferan genomes have been published to date, two species of chitons (Polyplacophora; Varney et al. 2021; Varney and Yap-Chiongco et al. 2022) and one species of Caudofoveata (Wang et al. 2024). Therefore, we sequenced the genomes of two species of Solenogastres – Neomenia megatrapezata Salvini-Plawen & Paar-Gausch 2004 and Epimenia babai Salvini-Plawen, 1997 – to facilitate further study of this evolutionarily important taxon.
The two clades of aplacophoran molluscs are united by their distinctive vermiform shape, loss or reduction of the foot, and a chitinous cuticle covered by calcareous integumental spines and scales (sclerites). Solenogastres are distinctive with the presence of a ciliated foot restricted to a ventral groove, an undifferentiated gut, and the lack of true ctenidia, while members of Caudofoveata lack a foot entirely and have the presence of a specialized oral shield used for burrowing in mud (Todt 2013). The current taxonomy of Solenogastres divides the group into 24 families and four orders (Pholidoskepia, Cavibelonia, Sterrofustia, and Neomeniamorpha) although recent phylogenomic analyses have indicated the need for significant taxonomic revision within this group (Kocot et al. 2019; Yap-Chiongco et al. 2024).
Whereas most solenogasters are just a few millimeters in length (Todt 2013) both N. megatrapezata and E. babai are large enough to obtain high-quality genomic and transcriptomic data from a single individual. E. babai is a species of Cavibelonia that is relatively easily collected at SCUBA-accessible depths where it feeds on the soft coral Scleronephthya gracillima (Okusu 2002). Because of its relatively large-body size (up to 20 cm in length) and reliable spawning within the laboratory, E. babai remains one of very few species of Solenogastres in which development has been described (Baba 1938, 1940, 1951; Okusu, 2002; Todt and Wanninger 2010). Sequencing of its genome coupled with characterized development and reliability of spawning make E. babai a desirable species to serve as a developmental model within Solenogastres. N. megatrapezata (up to 18 cm in length) is a species of Neomeniamorpha found in the Southern Ocean off Antarctica (Salvini-Plawen & Paar-Gausch 2004; Kocot et al. 2019). Neomenia is interesting to study trait evolution within Solenogastres as species within this genus have a complex accessory genital apparatus and lack both ventral foregut glands and a radula (García-Álvarez & Salvini-Plawen 2007). Together, the sequencing of these two species expands our ability to explore solenogaster evolution and, more broadly, provides an important resource in identifying large-scale patterns in molluscan evolution.
Epimenia babai was collected by Akiko Okusu as described in Okusu (2002). The specimen of Neomenia megatrapezata (collector number Ap227) used for genome and transcriptome sequencing was collected by Ken Halanych and Kevin Kocot on 3 February 2013 near the Ross Ice Shelf in Antarctica (NBP12-10 Station 22; 76° 59.8965′ S, 175° 05.5920′ W, 541 m depth) via Blake trawl and is deposited in the Alabama Museum of Natural History (ALMNH) under catalog number ALMNH:Inv:25736.
Additional solenogasters were used for transcriptome sequencing to provide evidence for genome annotation. A second individual of N. megatrapezata (Ap259) was collected by Ken Halanych and Kevin Kocot on 28 November 2013 near Recovitza Island, Antarctica (LMG13-12 Station 2; 64° 24.672′ S, 61° 57.790′ W, 664 m depth) via Blake trawl. A second individual from the same collection event has been deposited in ALMNH under catalog number ALMNH:Inv:25732 as a voucher. A specimen of Neomenia aff. herwigi (Ap20; same individual used to generate a 454 transcriptome by Kocot et al. 2011; NCBI SRA accession number SRR108985) was collected by Ken Halanych on 6 December 2004 off Argentina (63°23′ 03.0″ S, 60° 03′ 24.0″ W, 277 m depth) via Blake trawl. A specimen of Neomenia permagna (Ap26) was collected on 15 May 2006 by Ken Halanych off Argentina (LMG06-05; 53°47′ S, 60°42′ W; 170 m depth) via Blake trawl and is deposited in ALMNH under catalog number ALMNH:Inv:25733. A specimen of an unidentified species of Epimenia (possibly also E. babai) was collected on 29 June 2015 by Hiroshi Saito off Osezaki, Japan (55 m depth) by SCUBA diving and a tissue sample is deposited in ALMNH under catalog number ALMNH:Inv:25734.
To produce short-read genomic data, DNA was extracted using the CTAB-phenol-chloroform method employed by Varney et al. (2021), which was based on Doyle & Doyle (1991). For E. babai, DNA was extracted from a piece of midbody tissue and sent to Iridium Genomics for Illumina TruSeq sequencing library preparation and 2 X 100 bp paired-end (PE) sequencing on an Illumina HiSeq X. For N. megatrapezata, DNA was extracted from hemolymph and sent to the NY Genome Center for Illumina TruSeq sequencing library preparation and sequencing on an Illumina HiSeq X.
To produce long-read data via Oxford Nanopore sequencing, a library for N. megatrapezata was produced from DNA extracted from haemolymph using a CTAB-phenol-chloroform method following Varney et al. (2021). For the three libraries for E. babai and the second library for N. megatrapezata, genomic DNA was extracted from cryo-preserved tissue using the EZNA Tissue DNA Kit (Omega Bio-tek) followed by cleaning and size selection for high-molecular-weight fragments with Ampure XP beads. Sequencing libraries were prepared with the LSK-109 ligation-based library preparation kit and sequenced in-house using R9.4.1RevD flow cells on a GridION. Reads were base called with Guppy 4.0 and trimmed with PoreChop (Wick 2018) with the --discard_middle flag.
New transcriptome data were generated from the same individual used for WGS for each species and from other individuals of the same or closely related species for genome annotation. For E. babai, Epimenia sp., Neomenia aff. herwigi, and Neomenia permagna, RNA was extracted as described by Kocot et al. (2019). RNA concentration was measured using a Qubit 3.0 (Thermo Fisher) fluorometer with the RNA High Sensitivity kit, RNA purity was assessed by measuring the 260/280 nm absorbance ratio using a Nanodrop Lite (Thermo Fisher), and RNA integrity was evaluated using a 1% SB agarose gel. Complementary DNA (cDNA) synthesis was performed using 1 ng of total RNA using the Clontech SMART-Seq HT kit. An Illumina sequencing library was then prepared in house using the Nextera XT DNA Library Preparation Kit with 0.15 ng of cDNA. Final library size and concentration were assessed using an Agilent Fragment Analyzer with the NGS 1-6000 bp kit and sent to Psomagen (Cambridge, MA) for sequencing on an Illumina NovaSeq using a S4 flowcell with 2 X 150 bp PE reads. For N. megatrapezata, RNA was extracted from the individual used for genome sequencing (Ap227) and a second individual (Ap259) as described in Kocot et al. (2019) and sent to the High-Throughput Genomics Shared Resource of the Huntsman Cancer Institute at the University of Utah for Illumina TruSeq Stranded mRNA Sample Prep Kit with oligo (dT) selection. Sequencing was performed on the Illumina HiSeq 2500 system with 2 X 125 bp PE reads.
Genome size and heterozygosity were estimated based on trimmed reads using GenomeScope2 (Ranallo-Benavidez et al. 2020) with a k-mer of 21. Hybrid genome assembly was performed with MaSuRCA 3.3.5 (Zimin et al. 2017). Recommended settings for eukaryotes with >20X Illumina coverage were used. Genome quality was assessed following assembly (and after each step involving filtering or polishing the genome assembly; see below) with QUAST 5.0.2 (Mikheenko et al. 2018) and completeness with BUSCO 5.2.2 (Manni et al. 2021) using the Metazoa odb_10 dataset and the “--long” flag. We then removed redundant haplotigs with Redundans (Pryszcz & Gabaldón 2016) using the flags --noscaffolding --norearangements. Finally, the remaining scaffolds were polished with four rounds of Pilon 1.23 using the Illumina paired-end reads, which were first quality- and adapter-trimmed with trimmomatic 1.8.0 (Bolger et al. 2014) using “ILUMINACLIP:adapters.fasta:2:30:10 LEADING 10 TRAILING 10 SLIDINGWINDOW:4:15 MINLEN:50” for N. megatrapezata and “ILLUMINACLIP:adapters.fasta:3:30:10 LEADING:10 TRAILING:10 SLIDINGWINDOW:4:20 MINLEN:50” for E. babai.
For genome annotation, we followed a bioinformatic pipeline that masked repetitive DNA and leveraged both RNA-seq data from that species and protein sequences from diverse aplacophorans as evidence for gene modeling. Repeats in the final assemblies were annotated and softmasked with RepeatMasker using a custom repeat database generated with RepeatModeler (Smit & Hubley 2015). For RepeatModeler, a maximum genome sample size of 1M and the --LTRStruct option were used. For RepeatMasker, the slow and gccalc options were used. The engine used for both programs was rmblast. Available aplacophoran and select other mollusc proteomes (see data on Figshare for details) were then aligned to the final genome assemblies with ProtHint 2.6 (Brůna et al. 2020) with an e-value cutoff of 1e-25. We ran TrimGalore (Krueger et al. 2021) on the transcriptome reads with the following settings: “-q 30 --illumina --trim-n --length 50.” The trimmed and filtered transcriptome reads were then mapped to the genome using STAR 2.4.0k (Dobin et al. 2013) with “--genomeChrBinNbits 15 --chimSegmentMin 50.” Annotation of protein-coding genes was performed with BRAKER 2.1.6 (Brůna et al. 2021) using the output of ProtHint and STAR with the following settings: “--eptmode --softmasking --crf.”
Homologous protein sequences in our newly sequenced solenogaster genomes and the proteomes of 21 other lophotrochozoans, including 16 other molluscs, two annelids, one brachiopod, one phoronid, and one nemertean were used as input for orthology inference using OrthoFinder 2.4.0 (Emms & Kelly 2019). We then further refined the output of OrthoFinder to construct a matrix of one-to-one orthologs following the pipeline of Varney and Yap-Chiongco et al. (2022) except we retained only genes sampled for 18/23 taxa using PhyloPyPruner (https://gitlab.com/fethalen/phylopypruner). Maximum likelihood phylogenetic analysis was employed on the concatenated supermatrix of amino acid sequences in IQ-Tree 2.1.3 (Minh et al. 2020) using the best-fitting model for each partition (-m MFP) with 1000 rapid bootstraps. The tree was arbitrarily rooted with all non-molluscan taxa. Notably, as the genome of Chaetoderma sp. (Wang et al. 2024) was released following the completion of this analysis, it was not included.
For N. megatrapezata, Illumina genome sequencing yielded 443.02 M reads. Illumina transcriptome sequencing of the same individual of N. megatrapezata used for genome sequencing (Ap227) yielded 29.72 M reads and 32.93 M reads for a second individual of N. megatrapezata (Ap259). Transcriptomes of other Neomenia species resulted in 110.68 M reads for N. aff. herwigi and 24.40 M reads for N. permagna. GenomeScope analysis estimated the genome size of N. megatrapezata to be 294 Mbp and a heterozygosity of 0.142% based on the trimmed PE reads. Following adapter trimming, two flowcells of Oxford Nanopore sequencing yielded 101.31 M reads. Assembly with MaSuRCA resulted in an assembly of 9,870 contigs totaling 347 Mbp (N50 = 119 Kbp, L50 = 1,041) with a BUSCO completeness score of 87.1% (83.9% single, 3.2% duplicated, 8.0% fragmented, and 4.9% missing). After polishing and purging redundant haplotigs, the final N. megatrapezata assembly was reduced to 6,168 contigs totaling 412 Mbp (N50 = 132 Kbp, L50 = 881) with a BUSCO completeness score of 85.1% (81.7% single, 3.4% duplicated, 8.1% fragmented, and 6.8% missing). BRAKER predicted 65,328 gene models with 90.6% of BUSCOs detected (80.5% single, 10.1% duplicated, 4.5% fragmented, and 4.9% missing). Removal of gene models not supported by transcriptome or protein evidence resulted in a final gene set for N. megatrapezata of 22,463 with a BUSCO completeness score of 90.2% (81.0% single, 9.2% duplicated, 4.7% fragmented, and 5.1% missing).
For E. babai, Illumina PE sequencing yielded 693.24 M reads. Illumina transcriptome sequencing of E. babai and Epimenia sp. yielded 70.06 M and 131.47 M reads, respectively. GenomeScope analysis of PE reads failed to converge for E. babai with k-mer sizes of 17 and 21. Therefore, we used the trimmed nanopore data in the program KMC 3 (Kokot et al. 2017) with the options -k21 -fm -t16 -m500 -ci1 -cs10000 to obtain k-mer statistics followed by kmc-tools with the transform option to create a histogram for input into GenomeScope2. Oxford Nanopore sequencing of three flowcells data yielded 14.79M reads post adapter trimming with PoreChop. The resulting analysis based on Nanopore data estimated a genome size for E. babai of 543 Mbp and heterozygosity of 1.16%. MaSuRCA yielded an assembly of 11,695 contigs totaling 572 Mbp (N50 = 271 Kbp, L50 = 618) with a BUSCO completeness score of 91.3% (77.7% single, 13.6% duplicated, 5.2% fragmented, and 3.5% missing). The final polished and purged Epimenia assembly was reduced to 5,965 contigs totaling 628 Mbp (N50 = 413 Kbp, L50 = 370) with a BUSCO completeness score of 90.1% (82.0% single, 8.1% duplicated, 6.0% fragmented, and 3.9% missing). BRAKER predicted 123,904 gene models with a BUSCO completeness score of 93.3% (80.9% single, 12.4% duplicated, 4.7% fragmented, and 2.0% missing). Removal of genes not supported by transcriptome or protein evidence resulted in a final gene set for E. babai of 25,393 with a BUSCO completeness score of 92.4% (80.5% single, 11.9% duplicated, 4.9% fragmented, and 2.7% missing).
Molluscan genomes sequenced to date range in size from 359 Mbp (Simakov et al. 2013) to over 6 Gbp (Song et al. 2023) and genome size estimates by Adachi et al. (2021) for 141 diverse molluscs (Polyplacophora, Gastropoda, Bivalvia, and Cephalopoda) based on flow cytometry ranged from 469.4 Mbp (measured as a C-value of 0.48) to 5.31 Gbp (C-value of 5.43) with a mean size of 2.61 Gbp (C-value of 2.67) among the sampled molluscan taxa. Kocot et al. (2016) utilized Feulgen image analysis densitometry (FAID) to estimate genome sizes in select species of aplacophorans, including the same individual of Epimenia babai sequenced here. Genome size of E. babai was estimated to be 750 Mbp and that of Neomenia permagna was estimated to be 293 Mbp (Kocot et al. 2016). Genome size for E. babai inferred by GenomeScope based on Oxford Nanopore data is smaller than expected based on FIAD at 543 Mbp. However, we were unable to obtain an estimate based on more accurate PE data as the analysis failed to converge. The estimated genome size for N. megatrapezata of 294 Mbp inferred by GenomeScope is comparable to the FIAD estimate of 293 Mbp for the closely related species N. permagna. With final assembly sizes of 628 Mbp and 412 Mbp for E. babai and N. megatrapezata, respectively, solenogaster genomes are fairly small with respect to other molluscs. Based on computational and FAID genome size estimates, genome size within Aculifera is quite variable ranging from as small as 293 Mbp in N. permagna to 2.84 Gbp in Cryptochiton stelleri (Kocot et al. 2016; Hinegardner, 1974). Comparing the aplacophoran classes, the estimated genome size of Chaetoderma sp. (Caudofoveata) is quite large (2.45 Gbp) in comparison to both E. babai and N. megatrapezata (Table 1). Although in some groups it has been shown that genome size may co-evolve with life-history traits, the observed high level of variation in genome size within Aculifera raises questions about the underlying mechanisms driving this heterogeneity in genome size (Ritchie et al. 2017; Beaudreau et al. 2021). Further comparisons between solenogaster, caudofoveate, and chiton genomes will aid in our understanding of the evolutionary history of Aplacophora and Aculifera and the potential interplay between genome size and, e.g., life history.
| Species | Class | Size (Gbp) | N50 (Mbp) | BUSCO complete | # Genes | Citation |
|---|---|---|---|---|---|---|
| Epimenia babai | Solenogastres | 0.63 | 0.413 | 90.1 | 25,393 | This study |
| Neomenia megatrapezata | Solenogastres | 0.41 | 0.132 | 85.10% | 22,463 | This study |
| Chaetoderma sp. | Caudofoveata | 2.45 | 141.46 | 89.52% | 23,675 | Wang et al. 2024 |
| Acanthopleura granulata | Polyplacophora | 0.61 | 23.9 | 97.40% | 81,691 | Varney et al. 2021 |
| Hanleya hanleyi | Polyplacophora | 2.52 | 0.065 | 92.00% | 69,284 | Varney and Yap-Chiongco et al. 2022 |
| Liolophura japonica | Polyplacophora | 0.61 | 37.34 | 96.10% | 28,010 | Hui et al. 2024 |
Comparison of the full set of Epimenia and Neomenia gene models to the gene models from 21 other lophotrochozoans in OrthoFinder resulted in 251,468 groups of homologous sequences. Our pipeline selected 7,561 single-copy genes sampled for at least 18 of the 23 taxa. The final PhyloPyPruner output resulted in a supermatrix of 3,577 genes with a total of 1,169,375 amino acids and 20.3% missing data. Of these, E. babai was sampled for 2,319 and N. megatrapezata was sampled for 2,232.The resulting tree (Figure 1) is strongly supported overall with maximal support for Solenogastres and for Aculifera as the sister taxon to all other sampled molluscs, consistent with recent studies (e.g., Kocot et al. 2020; Song et al. 2023).
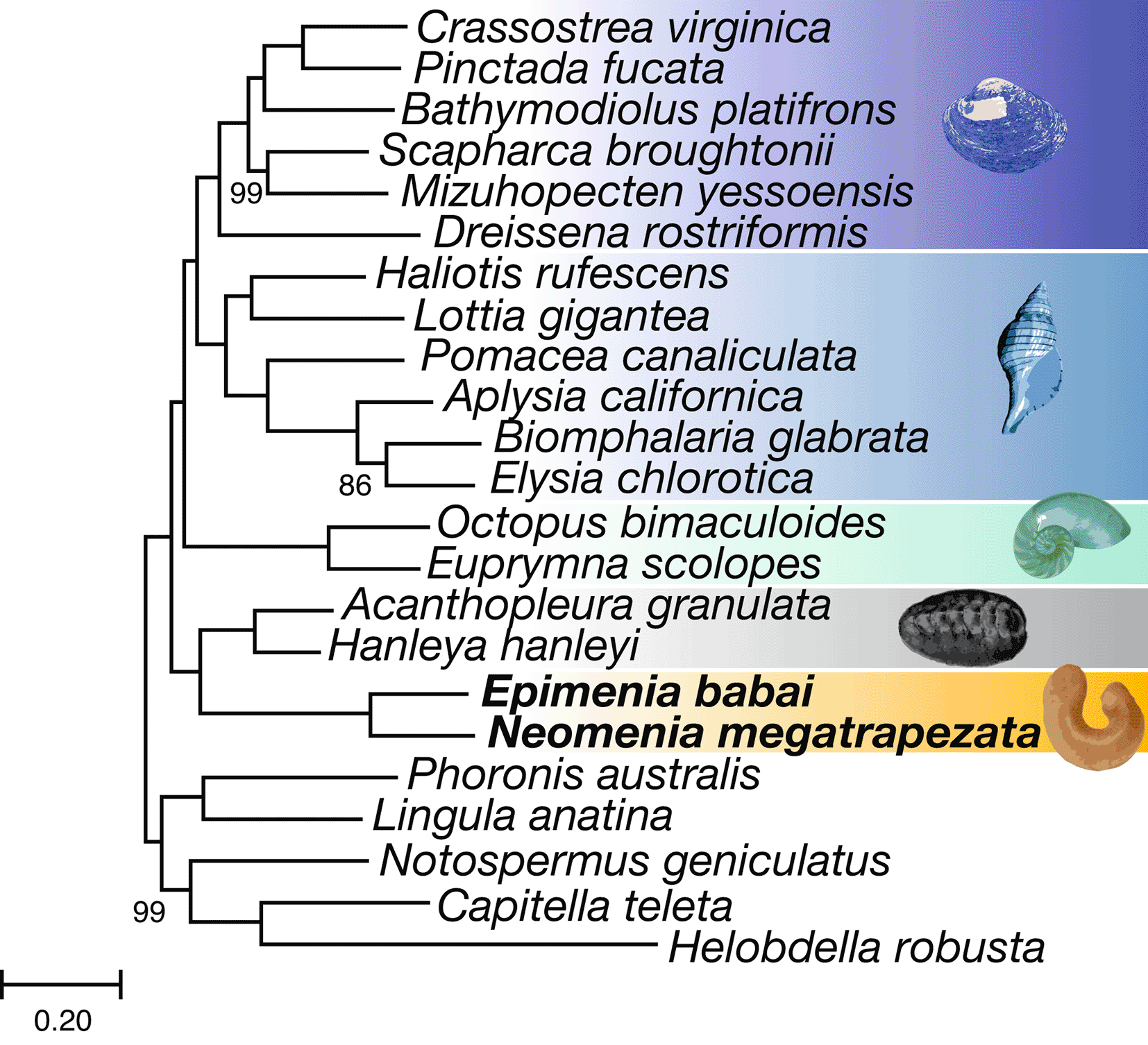
Bootstrap support values below 100 are displayed at each node. Image of Neomenia megatrapezata was altered from a photo taken by Christoph Held.
In summary, the genomes of N. megatrapezata and E. babai are relatively complete and comparable to other published aculiferan genomes (Table 1) with BUSCO completeness scores of 85.1% and 90.1%, respectively. These represent the first sequenced genomes for Solenogastres and are a valuable resource for future studies of molluscan evolution.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)