Keywords
Tethya wilhelma, Tethya minuta, Porifera, Demospongiae, genome, model organism
This article is included in the Genomics and Genetics gateway.
This article is included in the Nanopore Analysis gateway.
Tethya wilhelma, Tethya minuta, Porifera, Demospongiae, genome, model organism
We would like to thank the two reviewers for their valuable and constructive comments and their appreciation of the quality of our work.
We have attended all their comments, the majority of which were minor changes, except the request for a revised Figure 2. This Figure has been reworked accordingly and more information is now provided in the figure caption for an improved understanding. A detailed response to each comment by the reviewers has been provided.
See the authors' detailed response to the review by Eric Bautista-Guerrero
See the authors' detailed response to the review by Bernie Degnan
Sponges (Phylum Porifera) are sessile aquatic metazoans that occur globally in marine and freshwater habitats. More than 9,600 valid species have been described, the majority of which (7,989 species) belong to Class Demospongiae.1 Sponges hold a pivotal position in the animal tree of life as one of the earliest metazoan branching lineages, likely originating more than 650 million years ago,2 but their exact phylogenetic position is still disputed.3–6 As filter feeders, sponges are ecologically important, especially in coral reefs,7,8 but are also impacted by climate change, as they bleach due to elevated seawater temperatures.9,10 In the current biodiversity crisis, genome data is valuable to aid species conservation11 and genomic data can be used not only to understand evolution and development,12 but also to predict a species evolutionary potential to adapt to changing environmental conditions due to climate change.13 To improve the understanding of their response to changing environmental conditions, the availability of culturable sponge species with annotated high-quality genomes is important, but only a few sponge species meet both these criteria yet.
Although the first sponge genome was published in 2010 from the Australian demosponge Amphimedon queenslandica,14 only a handful of annotated high-quality sponge genomes have been published and analysed since then, for example from the freshwater demosponge Ephydatia muelleri15 or the reef-building glass sponge (Class Hexactinellida) Aphrocallistes vastus.16 However, only A. queenslandica and E. muelleri are culturable yet under controlled conditions. This lack of high-quality genome resources available from culturable sponges hinders a full appreciation of our understanding of animal evolution and function as well as the response of sponges as ecological key players in many aquatic ecosystems to the current climate crisis.
To contribute to filling this gap, we here provide high-quality draft genomes of the two aquarium species Tethya wilhelma and Tethya minuta. These two congeners are small ball-shaped demosponges that were described in 2001 from public aquaria in Germany.17 Due to their long-term culturability, they are a laboratory model for many topics, including multicellularity, early-animal evolution, biomineralization, and even cancer (e.g., Refs. 18–25). With the provision of novel high-quality genome data of these two species we aim to enhance the use of these species as valuable sponge model systems.
Specimens of Tethya wilhelma and Tethya minuta were obtained from the marine research aquaria of the Chair of Paleontology and Geobiology of the Department of Earth and Environmental Sciences at the Ludwig-Maximilians-Universität München (Germany), where they are cultured since about 2010. No permits were needed for the sampling and processing. Voucher specimens are deposited in the Bavarian State Collection for Paleontology and Geology (SNSB-BSPG) under accession numbers SNSB-BSPG.GW33333 (T. wilhelma) and SNSB-BSPG.GW41624 (T. minuta).
For both species, genomic DNA was extracted from either fresh or frozen tissue with a modified cetyltrimethylammonium bromide (CTAB; Carl Roth, Germany, Cat. Nr. 9161.1) extraction. The modification concerned the addition of Potassium acetate (KOAc, Sigma-Aldrich, Germany, Cat. Nr. 791733) in step no. 5 of the protocol.26 Short DNA fragments were removed using AMPure XP (Beckman Coulter, USA, Cat. Nr. A63881) beads to select for long DNA fragments. DNA quantity and quality were controlled on a Nanodrop 1100 and using 1.5% agarose (Biozym, Germany, Cat. Nr. 840004) gels before library preparation as required for the different sequencing platforms used.
For Tethya wilhelma, we took the genome draft (T. wilhelma-v1) published by our group in 201720 as a starting point and used new sequence data and bioinformatics to further improve it. Additional data was obtained using Hi-C27 and Chicago (Dovetail Genomics28) libraries. For this, four whole sponges were frozen in liquid nitrogen and shipped to Dovetail Genomics (Scotts Valley, CA) for library preparation and sequencing. The resulting Chicago/Hi-C reads were processed using Dovetail’s proprietary software HiRise.28 After Chicago/Hi-C scaffolding, the assembly (dubbed T. wilhelma-v2) had 1,353 scaffolds, totaling 139 Mb, with an N50 of 5.5 Mb.
While some chromosome-sized scaffolds were evident in T. wilhelma’s-v2 assembly, many putative chromosomes remained fragmented. Therefore, we tried to improve the assembly’s contiguity by adding Moleculo long reads as well as Nanopore long reads, the latter derived from a single run of an Oxford Nanopore MinION (see Table 1). The data was assembled using the programs “SSPACE_Long_Read v1-1”29 and “GapCloser v.1.12”.30 This assembly version, called Twi-v3, contained 967 scaffolds, totaling 138.92 Mb, with an N50 of 6.1 Mb (see Table 2).
All data can be accessed through the European Nucleotide Archive (ENA) https://www.ebi.ac.uk/ena/browser/view/.
| Version | # scaffolds | Total size (Mb) | N50 (Mb) | N gaps (kb) |
|---|---|---|---|---|
| Twi-v1 | 5936 | 125.67 | 0.073 | 1516 |
| Twi-v2 | 1353 | 139.49 | 5.5 | 2252.2 |
| Twi-v3 | 967 | 138.92 | 6.1 | 1069.9 |
| Twi-v4 | 891 | 138.93 | 6.7 | 1077.5 |
| Twi-v4-no_bacteria | 557 | 126.1 | 6.7 | 1030.7 |
For the assembly of T. wilhelma’s genome v4, high molecular weight DNA was extracted, and quality was assessed using a Nanodrop 1100. Fragment size was controlled on a 1.5% agarose gel and an Agilent 2200 TapeStation. Libraries for 10X Genomics (Pleasanton, CA) were generated and sequenced at the University of Potsdam, in collaboration with the group of Prof. M. Hofreiter (Evolutionary Adaptive Genomics, University of Potsdam, Germany), on an Illumina Nextseq500. About 390 M reads were obtained (Table 1) and assembled using the 10X-Genomics software “Supernova 2.1.1”.31 These assembled contigs were then used for scaffolding the T. wilhelma v3 assembly using “SSPACE-LongRead v1-1”29 and then with “P_RNA_scaffolder”32 using 100.7 M PE (125 bp) and 237 M RNA reads (25.2 Gb). Finally, we used hicstuff 2.3.033 and the Hi-C data available to create a contact map of the T. wilhelma assembly. This assembly, called Twi-v4, had 891 scaffolds with a total size of 138.9 Mb and an N50 of 6.7 Mb, which also included bacterial scaffolds (see Table 2 and below).
For the assembly of Tethya minuta, DNA of a single specimen of Tethya minuta (sample# GW41624) was extracted with CTAB26 and sequenced twice, using Oxford Nanopore PromethION (12.73 Gb of long reads) and MinION34 (2.17 Gb of long reads). Additionally, we Illumina-sequenced 27 Mbp paired-end (100 PE and 150 PE). These data were assembled with wtdbg2,35 and polished using minimap2.36 SSPACE_LongRead29 was used with the available nanopore data to scaffold the assembly. Finally, we used GapCloser 1.1230 and the available PE reads to close gaps in the assembly which then had a length of 139 Mb and consisted of 1,043 scaffolds (Tmi-v4), but still included bacterial contigs (see Table 3).
From earlier versions of the genome, it was clear that Tethya wilhelma harbours two associated bacteria, both alphaproteobacteria, with an unknown interaction. With the relatively large scaffolds in the assembly, a clear split was seen in GC content and read mapping coverage (Figure 1). Consequently, we separated all scaffolds with GC content under 47% and defined those as sponge. The remaining scaffolds for the two bacteria were binned using “MetaBAT v2.15-25”,37 with default parameters. For T. wilhelma, this yielded 6 bins (see public data repository at Ref. 38 or https://github.com/PalMuc/2Tethya_genomes/tree/main/03-bacteria) with 1 bin corresponding to a single Rhizobiales species (genome appx 7.5 Mb), and the other 5 bins corresponding to a Roseobacter species (genome appx 4.8 Mb). These scaffolds were removed from the final Tethya wilhelma genome version. This assembly, called Twi-v4-no_bacteria, had 557 scaffolds with a total size of 126.1 Mb and an N50 of 6.7 Mb (Table 2; see Ref. 38 or https://github.com/PalMuc/2Tethya_genomes/tree/main/06-FINAL_Assemblies ENA accession GCA_964030475).
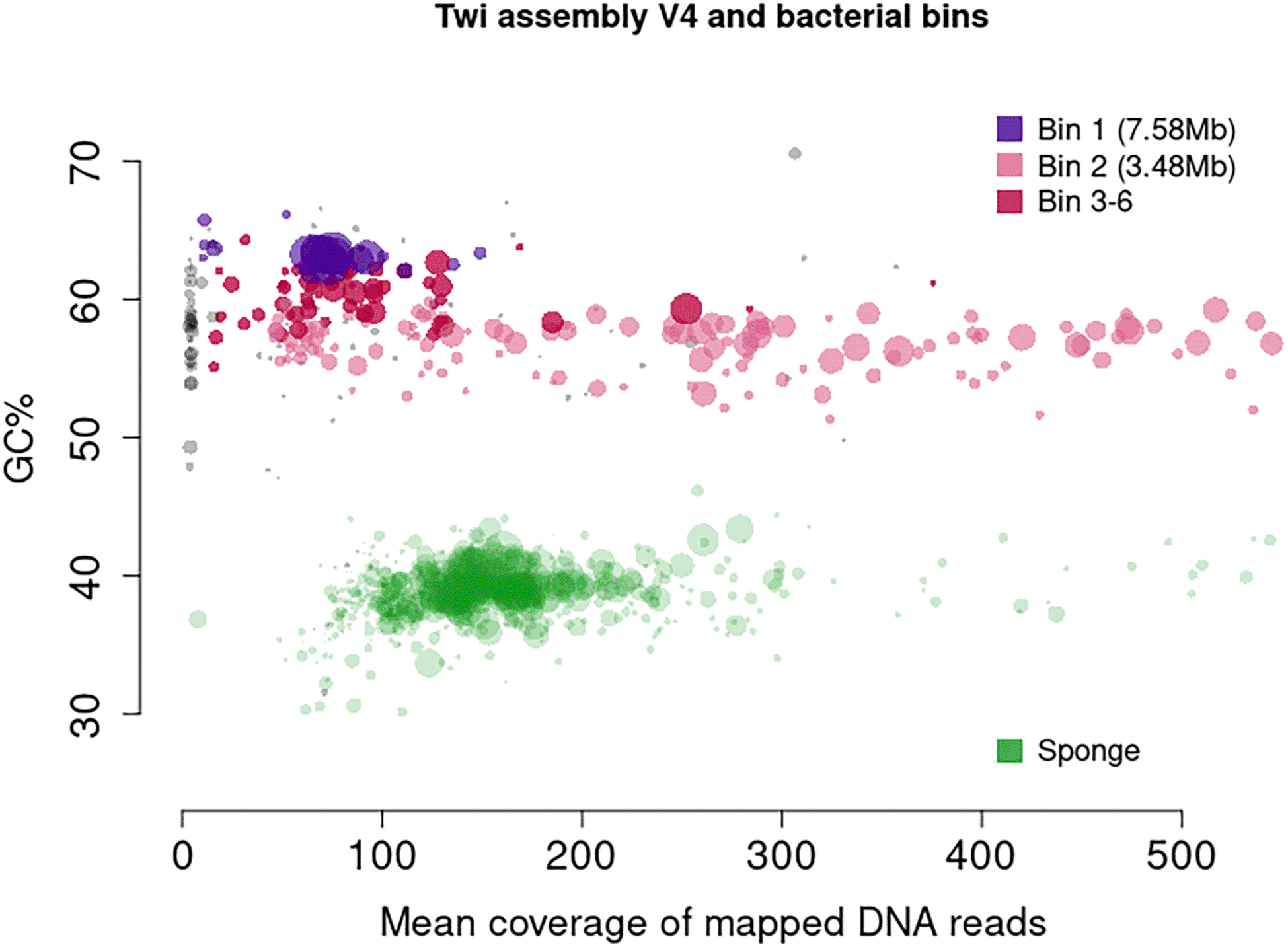
MetaBAT v2.15-25 was used for binning. Unidentified scaffolds are shown in grey.
For T. minuta, we used “MetaBAT v2.15-25”37 to identify and separate bacterial contigs from sponge scaffolds, which had produced 23 bins from the assembly. One of the bins, numbered as bin-17, contained the bulk of the assembly, and was identified as originating from the sponge due to the GC content of 38.3% and substantial RNAseq mapping. This bin (here now called Tmi-v4-no_bacteria, Table 3) had 244 scaffolds with a total size of 86.07 Mb, around 40 Mb smaller than the assembly of T. wilhelma (Table 2, see Ref. 38 or https://github.com/PalMuc/2Tethya_genomes/tree/main/06-FINAL_Assemblies; ENA accession GCA_964030485). Nearly all of the large chromosomal pieces in T. wilhelma had matching pieces among the scaffolds of T. minuta, as evident on the synteny plot (Figure 2), which suggested that the assembly of T. minuta was smaller not because of missing scaffolds or mis-assemblies, but merely from a smaller genome.
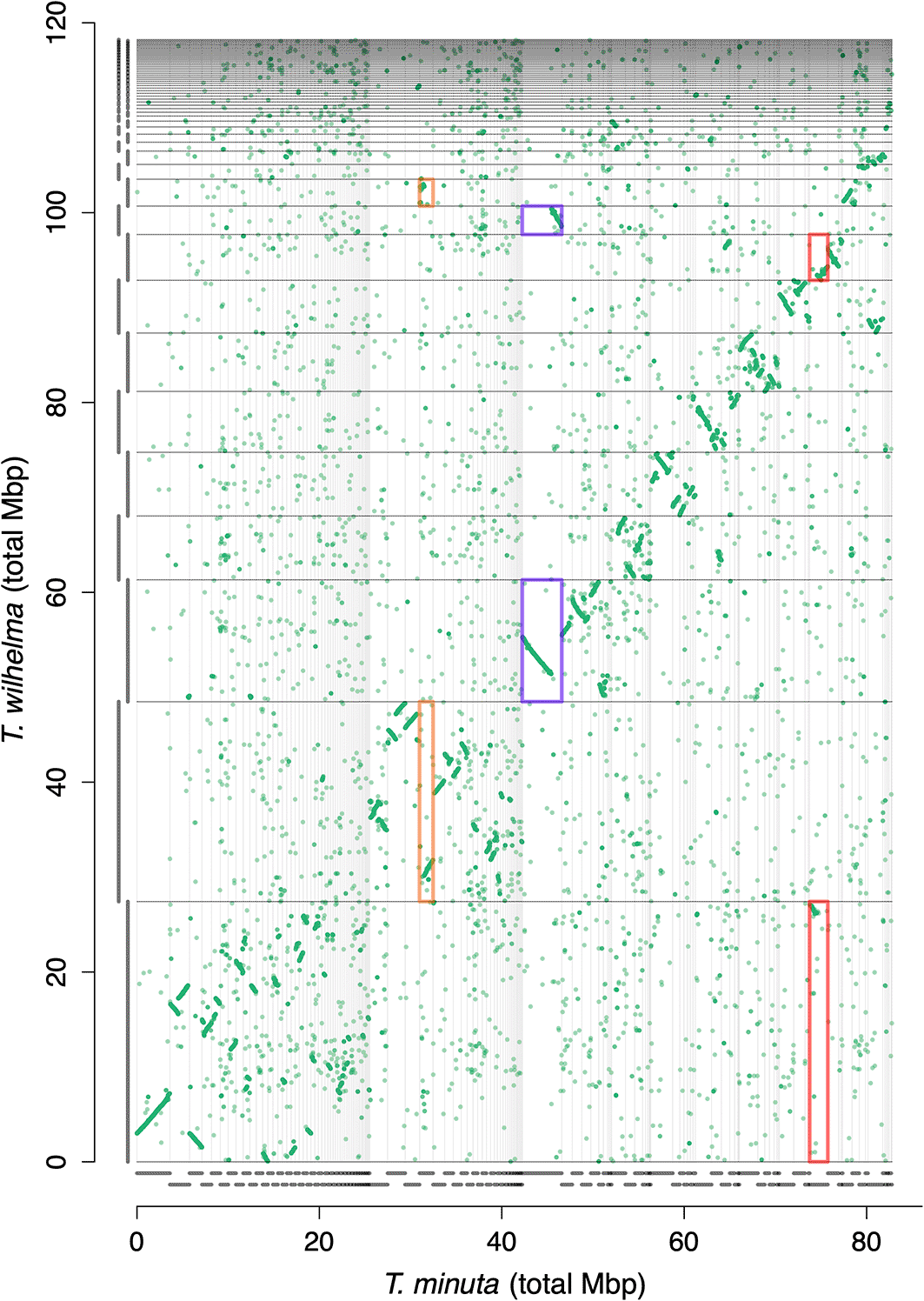
Each point represents a homologous gene between the two species. Bars parallel to each axis show the scaffold size. Scaffolds are arranged from longest to shortest in T. wilhelma, with the scaffolds in T. minuta sorted to match the T. wilhelma scaffold with the most homologs. Several rearrangements are evident, some are shown in the orange, blue, and red boxes, which could result from translocations, inversions on currently incomplete scaffolds, or misassemblies.
RNA was extracted from fresh tissue of Tethya minuta using TRIzol (Fisher Scientific, Germany, Cat. Nr. 12034977) and chloroform (Carl Roth, Germany, Cat. Nr. 3313.1) precipitation39 with subsequent quality control on a Bioanalyzer 2100. Libraries were prepared and sequenced twice using one third of a lane of an Illumina HiSeq1500 (100 bp and 50 bp) at the LMU GeneCenter, yielding 137 M read pairs. Reads were assembled de novo using Trinity,40 using default parameters, resulting in an assembly of 151,079 contigs with an average length of 677 bp. This assembly was also used as a training set for de novo gene prediction (see below). Transcriptome sequencing and assembly of Tethya wilhelma has been described in Francis et al.20 Statistics of the different sequencing libraries of Tethya wilhelma and Tethya minuta are given in Table 1.
Both Tethya species were annotated using AUGUSTUS. For T. wilhelma, we used the BRAKER v2.0 pipeline,41 with the options --useexisting --species=Tethya_wilh and including mapped RNA. This predicted a total of 28,113 gene models, which were used for downstream analysis.
For Tethya minuta, the assembly Tmi-v4-no_bacteria and the de novo Trinity assembly were used as inputs for WebAUGUSTUS.42 This yielded 22,779 gene models and 33,041 genes (see files ‘hints_pred’ and ‘hints_UTR_pred’ at Ref. 38 or https://github.com/PalMuc/2Tethya_genomes/tree/main/05-annotation/tethya_minuta_augustus).
The final version of the Tethya wilhelma draft genome assembly (see Table 2, Twi-v4-no_bacteria) without bacterial scaffolds has 557 scaffolds, a length of 126.1 MB, an N50 of 6.7 MB, and contains 1030.7 kb gaps (Ns). The final version of the Tethya minuta draft genome assembly (see Table 3, Tmi-v4-no_bacteria) without bacterial scaffolds has 244 scaffolds, a length of 86.07 MB, an N50 of 969.3 kb, and contains 534.5 kb gaps (Ns). BUSCO values for the two assemblies are given in Table 4.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)