Keywords
TTN, CTNNA3, dilated cardiomyopathy, pathogenic variants, familial segregation
This article is included in the Cell & Molecular Biology gateway.
TTN, CTNNA3, dilated cardiomyopathy, pathogenic variants, familial segregation
Dilated cardiomyopathy (DCM) is characterized by left ventricular dilatation and systolic dysfunction in the absence of significant coronary artery disease or volume overload. Although relatively rare in children, with an annual incidence estimated at 0.57 per 100,000, DCM is a major cause of heart failure and cardiac transplantation.1
In pediatric forms, the aetiology is often difficult to establish. Approximately 40% of childhood DCM cases remain idiopathic after standard investigations, but a growing number of studies have highlighted the significant role of genetic causes accounting for about 35% of cases.2,3 Over 50 genes have been implicated in the pathogenesis of dilated cardiomyopathy (DCM) to date.4,5 A number of these genes encode proteins essential for mitochondrial function and the preservation of cellular integrity, such as those coding for sarcomeric, cytoskeletal, nuclear envelope and Z-disk-associated proteins proteins.6,7
Among the most frequently implicated genes in familial DCM, the Tittin gene (TTN), encoding the sarcomeric protein Titin, is considered the main susceptibility gene in adults. It was reported that truncating variants in TTN (TTNtv) gene account for about 25% of non-ischemic DCM cases in adults.8,9 However, the contribution of TTN in pediatric cases is less defined, with a lower mutation rate and significant phenotypic variability.10
CTNNA3 gene, encoding alpha-T catenin, has been associated with inherited forms of arrhythmogenic right ventricular cardiomyopathy (ARVC). In a murine model, van Hengel et al. showed that CTNNA3 inactivation disrupted intercellular junction architecture and led to ventricular dilation.11
Whole-exome sequencing (WES) is increasingly emerging as a diagnostic tool, allowing the identification of rare and potentially pathogenic mutations even in the absence of a clear family history. The relevance of WES in clarifying complex cardiomyopathies and guiding familial screening has been reported.12,13
This study reports the clinical features of a severe DCM in a 2-year-old boy and links this pathogenesis with the co-existence of two pathogenic genetic variants in TTN and CTNNA3 genes. Overall, the identification of these mutations broadens the base of pathogenic variants linked to DCM.
A Tunisian family was referred to the Cardiology Department of Hedi Chaker Hospital in Sfax, Tunisia. Seven individuals of the family were enrolled in this study. Routine cardiovascular examinations, including electrocardiogram (ECG), cardiac echography, and cardiac magnetic resonance imaging (CMR) were performed.
Approval was granted by the Ethics Committee of the Faculty of Medicine of Sfax - Tunisia. Written informed consent was obtained from the parents of the proband and the participating family members.
Whole exome sequencing was performed on genomic DNA of the proband (IV-4). DNA was isolated using the QIAamp DNA Blood Mini Kit (Qiagen) following the manufacturer’s protocol. After enzymatic digestion, the target regions were enriched using DNA capture probes and the generated library was sequenced on an Illumina HiSeq 6000 platform, and aligned to the UCSC human genome GRCh37/hg19. Variants annotation was done using the BaseSpace Variant Interpreter and those with a minor allele frequency of less than 1% in the gnomAD database and disease-causing variants reported in HGMD and ClinVar databases are retained.
Variants identified in the proband by WES, were searched in available DNA samples extracted from family members. Regions including both variants were amplified by PCR using the following primers: for TTN: Forward: 5′- CGATTACACGGTGATCTGGT-3′; and Reverse: 5′-GCACTTTGACC ATGACAGACA-3′; and for CTNNA3: Forward: 5′-GGCTAAGATGACTCAACTGCC-3′, and Reverse 5′-TTGCTTTTCCCCTACCACCT-3′. Amplicons were sequenced using BigDye Terminator v3.1 Cycle Sequencing Kit and sequenced using the SeqStudio system (Applied Biosystems).
The pedigree of the family is shown in Figure 1. The patient is a 16-month-old boy who was admitted to the paediatric department for severe respiratory distress. He was born to non-consanguineous Tunisian parents who were reportedly healthy. He was the second child of the couple and he had an apparently healthy five-year-old brother. A detailed family history revealed that his grandmother died at the age of 56 years because of heart failure, his paternal aunt (patient III-1) was diagnosed with DCM at the age of fourteen and a cousin had died suddenly at the age of five months from unknown cause ( Figure 1). The perinatal history of the index case (patient IV-4) was unremarkable with a birth weight of 3000 g, length of 51 cm, Apgar score 9/10, and the psychomotor development was appropriate for age. He had experienced three episodes of bronchiolitis requiring hospitalization at two, four, and seven months of age, leading to a diagnosis of childhood asthma.
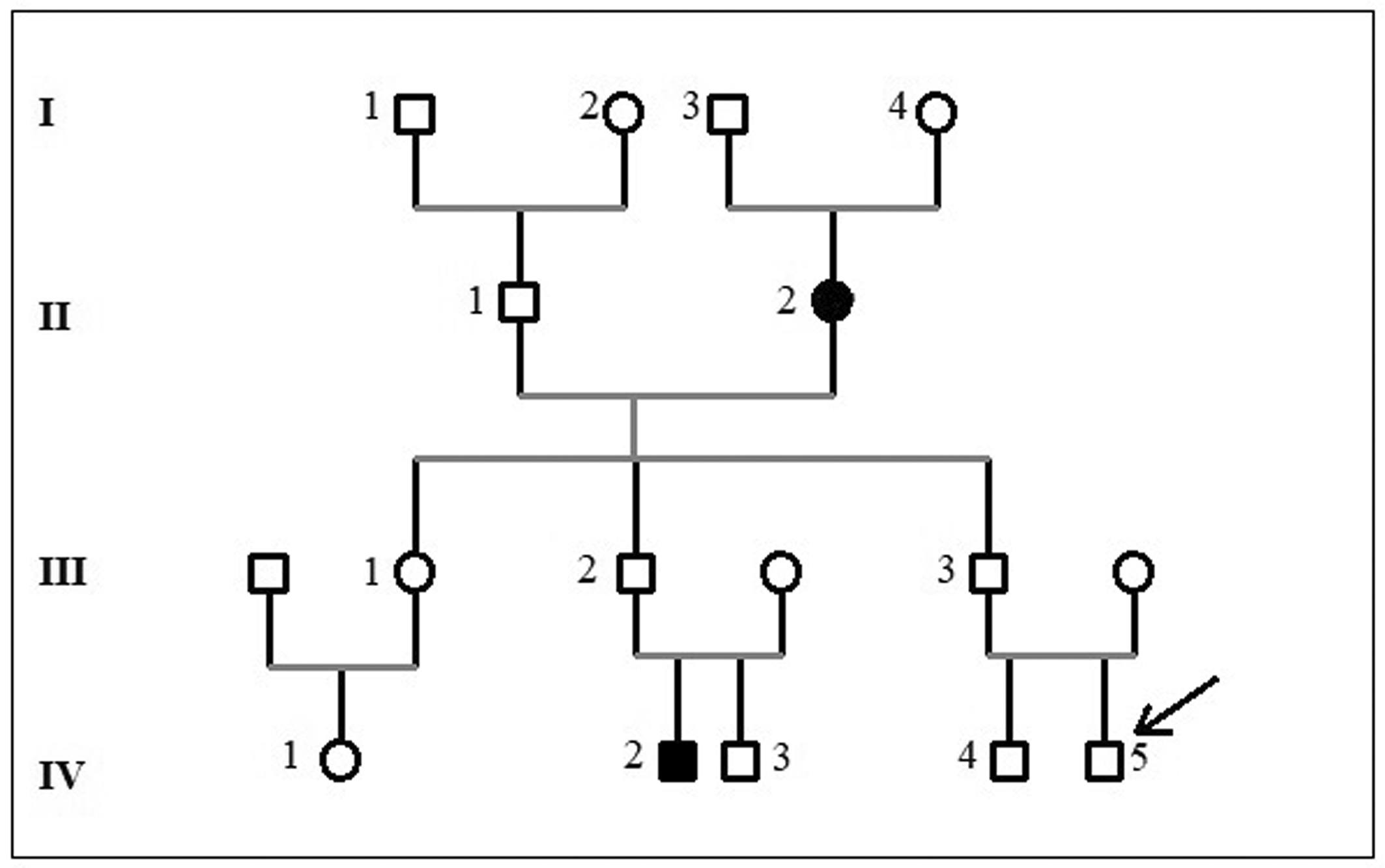
Males are shown as squares, and females are shown as circles. An arrow indicates the proband individual, and the black symbols indicate the deceased individual.
Clinical examination showed failure to thrive with a weight of 8 kg – 3DS a height of 50 cm (-2DS), blood pressure of 70/40 mmHg, heart rate of 165 beats/min, respiratory rate of 60 breaths/min with severe signs of heart failure and low cardiac output.
Laboratory investigations revealed elevated serum lactate level (3.2 mmol/L) with metabolic acidosis, high-sensitivity troponin level was elevated (80 ng/L). The ECG showed a sinus tachycardia without signs of myocardial ischemia. Chest radiography demonstrated cardiomegaly with pulmonary oedema. An echocardiogram revealed a severely dilated and hypokinetic left ventricle (LV) with a markedly decreased LV ejection fraction (EF) 15%, with reduced global longitudinal strain (–3.7%) and moderate mitral regurgitation secondary to annular dilatation ( Figure 2). Otherwise, cardiac architecture was normal, with no evidence of left ventricular outflow tract obstruction and a normal coronary artery pattern.
The proband was treated as follows: face mask oxygen (6 l/mn), furosemide (2 mg/kg/day) spirinolactone (3 mg/kg/day), thromboembolism prevention with acetylsalicylic acid (5 mg/kg/day) and dobutamine infusion (5 gamma/kg/min). Infectious workup was negative for common respiratory pathogens. Serological tests for cytomegalovirus and the Epstein-Barr virus were negatives.
Metabolic workups were also negative ruling out metabolic causes, including serum amino acids, urine organic acids, and L-carnitin level was in normal range: 52 μmol/L (normal values: 30-70 μmol/L).
The clinical course was marked by an improvement in heart failure symptoms, with progressive weaning from catecholamines, which allowed hospital discharge. He was readmitted to hospital two times after the initial episode at the age of 25 and 27 months respectively because of cardiac decompensation secondary to a pneumonia. He is now 27 month-year-old and he is listed for heart transplantation.
Furthermore, comprehensive echocardiographic evaluations were performed on additional family members, revealing varying degrees of left ventricular systolic dysfunction, as demonstrated by reduced global longitudinal strain (GLS) values in Figure 2.
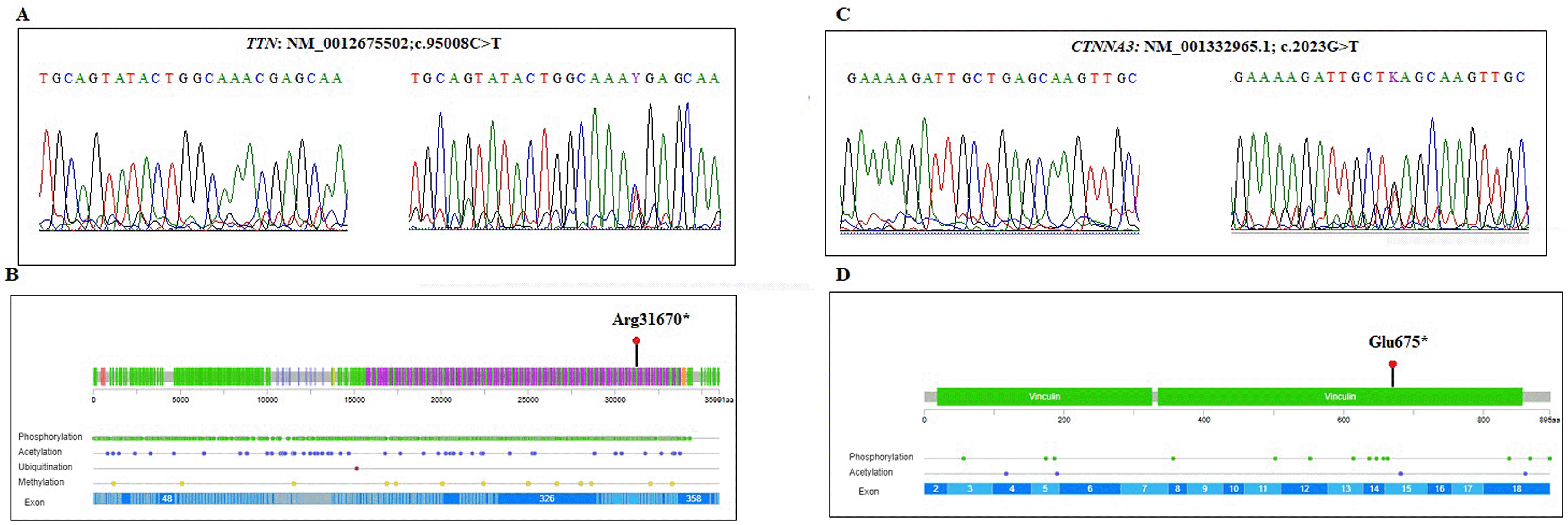
Whole Exome Sequencing (WES) was performed on genomic DNA extracted from the proband (IV-4). Through targeted data analysis of genes involved in cardiomyopathies, we identified two heterozygous germline variants namely a nonsense variant in exon 342 of the TTN gene (NM_001267550.2; c.95008C>T; p.Arg31670Ter) and a nonsense variant in exon 15 of the CTNNA3 gene (NM_001332965.1;c.2023G>T; p.Glu675Ter). Both variants were confirmed by Sanger sequencing ( Figure 3A) and are predicted to introduce premature stop codons, leading to truncated proteins that lack critical functional domains ( Figure 3B). The TTN variant (c.95008C>T) localized in the A-band coding the fibronectine III domain of the titin protein and has been reported previously as associated to DCM. This variant is rare, with a frequency of 8.21e-06 as reported in gnomAD v4 Exome. The PhyloP100way score of the c.95008C > T (p.Arg31670*) variant suggests the important evolutionary conservation of this site. According to the ACMG criteria (PVS1, PM2) the variant was classified as pathogenic/likely pathogenic. Furthermore, the CTNNA3 variant (c. 2023G>T) was novel and classified as likely pathogenic according to the ACMG criteria (PVS1, PM2). Segregation analysis within the family was conducted for the two identified variants across all available members. The results demonstrated that four individuals were compound heterozygous for both variants, two carried a single variant, and one was wild-type at both loci ( Table 1).
Dilated cardiomyopathy (DCM) is a clinically and genetically heterogeneous disorder characterized by dilation and impaired contraction of the left or both ventricles, leading to systolic dysfunction.1,14,15 Mutations in genes encoding sarcomeric and cytoskeletal proteins are major contributors to both familial and sporadic forms of DCM.5,16,17 Among these genes, TTN is the most frequently mutated in DCM.8,9
The TTN gene, encoding the sarcomeric protein titin, plays a critical role in maintaining myocardial elasticity and mechanical stability. Truncating variants in TTN (TTNtv) including nonsense mutations, frameshift mutations, and out-of-frame splice site variants, represent the most prevalent genetic cause of dilated cardiomyopathy (DCM).9 Studies on large cohorts reported that TTNtv were identified in 11-15% of adult cases of sporadic DCM and 23-27% of familial DCM.18–23
The pathogenic effect of TTNtv is position-dependent, with variants located in the A-band region of the gene being particularly deleterious, given their substantial disruption of sarcomeric structure and function.24
In this study, we identified a heterozygous nonsense variant, c.95008C>T (p.Arg31670*), in exon 342 of the TTN gene, which introduces a premature stop codon. This variant has been reported previously in familial forms of DCM.25 Familial segregation analysis confirmed that the variant co-segregated with the disease phenotype, being present in all affected individuals, while unaffected family members carried the wild-type allele.
The CTNNA3 gene encodes alpha-T catenin, a key component of the intercalated disc that plays a crucial role in myocardial intercellular adhesion.11 Although CTNNA3 has been less extensively studied in the context of DCM, it has emerged as a potential candidate gene.26,27 Notably, Janssen et al. proposed CTNNA3 as a candidate disease gene in a family with DCM linked to the 10q21–q23 locus, where this gene is located. However, no pathogenic variants were identified in that study, and the gene’s involvement in DCM remained inconclusive.28 Subsequent research demonstrated that alpha-T catenin protein levels are significantly reduced in DCM, and that decreased CTNNA3 expression correlates with left ventricular ejection fraction (LVEF) and ventricular dimensions, suggesting a potential role in disease progression.29 Recently, proteomic and phosphoproteomic approaches have provided evidence supporting a role for CTNNA3 phosphorylation in the pathophysiology of DCM.30
In this study, we identified a novel heterozygous nonsense variant in the CTNNA3 gene, c.2023G>T (p.Glu675*), in the proband. This variant introduces a premature stop codon and is classified as pathogenic based on ClinVar annotations and ACMG criteria.
Interestingly, both germline CTNNA3 and TTN variants coexist in the proband and other affected family members, suggesting a potential digenic contribution to the disease phenotype. Furthermore, two younger family members, who are currently asymptomatic, carried only one of the two identified variants either the TTN or the CTNNA3 nonsense pathogenic/likely pathogenic variant. This finding suggests the possibility of incomplete penetrance, variable expressivity, or age-dependent onset of the disease. Clinical evaluation, including transthoracic echocardiography, revealed no significant abnormalities in those individuals. The absence of a detectable phenotype in carriers of a single variant supports the hypothesis that co-inheritance of both TTN and CTNNA3 variants may be required for disease expression, consistent with a potential digenic inheritance model contributing to the pathogenesis of dilated cardiomyopathy in this pedigree. In our study, the early and severe disease onset at the age of 2 may reflect an additive or synergistic effect of the TTN and CTNNA3 variants, supporting the hypothesis that combined genetic hits may increase disease severity and accelerate its clinical manifestation.
The co-occurrence of pathogenic variants in cardiomyopathy-associated genes is relatively rare but has been increasingly recognized as a potential contributor to more severe or early-onset phenotypes. Recently, Han et al. report a case of DCM with a novel TTN variant, as well as two rare variants in the SCN5A and LDLR genes.31 It is interesting to mention that the TTN/CTNNA3 combination has not been reported previously in DCM.
This finding further underscores once again the genetic complexity of cardiomyopathy and highlights the utility of whole-exome sequencing (WES) in identifying novel candidate genes to improve patient management. In our previous work, we performed WES on a Tunisian family presenting with complex hypertrophic cardiomyopathy and identified a variant in the MRPL44 gene that segregates with the disease phenotype, suggesting a potential role for MRPL44 in the genetic etiology of hypertrophic cardiomyopathy.32
In conclusion, this case highlights the critical importance of comprehensive genetic evaluation in pediatric DCM, particularly when familial inheritance is suspected. The co-occurrence of two pathogenic variants in the TTN and CTNNA3 genes in a symptomatic family member underscores the genetic complexity of cardiomyopathies and supports the hypothesis of a multigenic model contributing to severe, early-onset phenotypes. These findings directly informed a targeted family screening strategy and prompted close cardiologic monitoring for at-risk relatives, enabling early identification and intervention.
The Institutional human ethics committee of the Faculty of Medicine of Sfax-Tunisia., approved this work (No65/24). Written informed consent to participate in this study was provided by all participants and, for children, by their legal guardian/close relative. All data were fully anonymized prior to analysis to ensure confidentiality, in compliance with ethical standards, and in adherence to the Declaration of Helsinki.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)