Keywords
Neotropical weevils; mitochondrial genome; COI barcode; Curculionidae; Passiflora foetida; biological control
This article is included in the Genomics and Genetics gateway.
Neotropical weevils; mitochondrial genome; COI barcode; Curculionidae; Passiflora foetida; biological control
Within Cryptorhynchinae subfamily (Coleoptera: Curculionidae), the Neotropical genus Philonis represents an underexplored lineage of stem-galling weevils that are tightly associated with species of Passiflora (Passifloraceae). Among them, Philonis inermis has recently attracted attention as a potential biological control agent against Passiflora foetida, an invasive vine that causes significant ecological and economic damage in Australia (Clavijo-Giraldo et al., 2025. Unpublished). Although a considerable number of cryptorhynchine species are known as agricultural pests, some species are being studied for their potential as biological control agents. P. inermis is one such example, exhibiting high host specificity towards P. foetida and inducing gall formation that weakens or kills the host plant. However, the lack of molecular data for Philonis hampers its accurate phylogenetic placement and limits the development of molecular tools for its identification and monitoring in both native and introduced ranges.
Mitochondrial genomes or mitogenomes have become fundamental tools for understanding the evolutionary history, systematics, and ecology of insects.1,2 In Coleoptera, the increasing availability of complete mitogenomes has facilitated robust phylogenetic analyses at various taxonomic levels, revealing patterns of diversification and adaptation in highly speciose families such as Curculionidae and other related weevil lineages.3–5 Despite these advances, mitogenomic data remain scarce for the subfamily Cryptorhynchinae, which encompasses a highly diverse assemblage of weevils with complex host-plant interactions.
Recent mitogenomic studies within Cryptorhynchinae have mainly focused on species of economic concern, particularly pests of woody plants and crops. Examples include Eucryptorhynchus chinensis and E. brandti, pests of Ailanthus altissima in China,6,7 and the mango seed weevils Sternochetus gravis, S. mangiferae, and S. olivieri.8 The hyperdiverse genus Trigonopterus, with hundreds of recently described species,9 also provides important comparative resources. Although related genera such as Aclees10 are not part of Cryptorhynchinae, they remain relevant for broader curculionid comparisons. In parallel with mitogenomic research, the use of the mitochondrial cytochrome c oxidase subunit I (COI) gene as a DNA barcode has become an essential tool for species identification, biodiversity assessment, and the detection of cryptic diversity in weevils and other insects.11,12 Despite its broad application in Curculionidae, COI barcoding data have been scarce for Neotropical Cryptorhynchinae, limiting our understanding of species boundaries and population structure in this group. Until now, no complete mitogenomes have been available for any Neotropical representative of Cryptorhynchinae, making P. inermis the first of its kind. The integration of mitogenomic and COI barcode data presented here provides a valuable reference for future studies on species delimitation, comparative genomics, and the development of molecular tools for monitoring and management of potential biological control agents.
In this context, the accurate delimitation of P. inermis is critical for any potential classical biological control strategy against P. foetida. Correct species identification, coupled with low intraspecific genetic variability, ensures the reliability and safety of introducing candidate agents into new environments. By combining complete mitochondrial genome sequencing with COI barcode analysis, this study not only provides the first comprehensive molecular characterization of P. inermis, but also demonstrates the utility of COI as a diagnostic marker for its unambiguous identification. These resources will serve as a foundation for both fundamental studies on Cryptorhynchinae evolution and applied research aimed at evaluating P. inermis as a safe and effective biocontrol agent.
This study aims to generate the first shadow-sequencing based genome to characterizing the complete mitochondrial genome of P. inermis and integrate it with COI barcode sequences to refine the molecular identification and comparative analysis of this species within Cryptorhynchinae. Through comparative analyses with available mitogenomes of related genera (Eucryptorhynchus, Sternochetus, and Trigonopterus), we explore patterns of gene arrangement, nucleotide composition, codon usage, and control region variability. These results not only fill a significant gap in the mitogenomic data of Neotropical Cryptorhynchinae but also provide essential molecular resources for evaluating P. inermis as a candidate biological control agent of P. foetida, linking fundamental evolutionary insights with applied biocontrol strategies.
Field surveys were conducted between 2021 and 2024 across dry forest habitats of northern Colombia (departments of Antioquia, Córdoba, and Bolívar; 0–200 m a.s.l.) to locate populations of P. foetida (Passifloraceae) exhibiting stem galls induced by P. inermis. The host plant was identified by botanist Wilder Buitrago Arbeláez (Herbarium of the Universidad de Antioquia, HUA, Medellín, Colombia) based on vegetative and floral characters following the diagnostic treatment reported elsewhere.13 A voucher specimen of P. foetida was deposited at the HUA Herbarium under collection number HUA-1633.
Gall-bearing stems were excised using sterile scissors, placed in individual 50 mL sterile polypropylene tubes (Falcon, Cat. No. FALC-352070X25), and transported to the Entomology Laboratory of Universidad Nacional de Colombia (Medellín). Each gall was incubated separately under controlled environmental conditions (25 ± 2 °C, 70 ± 5% relative humidity, 12:12 h light: dark cycle) until adult emergence to minimize contamination and sample mixing.
Adults were either preserved in 96% ethanol (Merck, Cat. No. 100983) for molecular work or mounted as pinned specimens for morphological examination. Species identification was confirmed through external morphological traits (rostrum length and curvature, elytral scale pattern, and sexual dimorphism) and dissection of male genitalia using a Leica EZ4 HD stereomicroscope (Leica Microsystems, Germany). Identification followed the diagnostic criteria of O’Brien14 and comparisons with authenticated reference material. Voucher specimens were deposited in the Francisco Luis Gallego Entomological Museum (Universidad Nacional de Colombia, Medellín) under catalog numbers NC 65188–NC 65220.
Genomic DNA was extracted from approximately 25 mg of thoracic muscle from ethanol-preserved adults using the DNeasy Blood & Tissue Kit (Qiagen, Germany; Cat. No. 69504) according to the manufacturer’s protocol. Each extraction used 180 μL Buffer ATL and 20 μL Proteinase K, with overnight digestion at 56 °C, followed by standard purification and elution in 100 μL Buffer AE.
DNA concentration and purity were quantified using a NanoDrop 2000/2000c spectrophotometer (Thermo Fisher Scientific, USA; Cat. No. ND-2000) and verified by 1% agarose gel electrophoresis in 1× TAE buffer (Invitrogen, USA; Cat. No. 15558-042) with GelRed stain (Biotium, USA; Cat. No. 41003). Only samples with concentrations ≥ 1 ng μL−1 and 260/280 and 230/260 ratios between 1.8 and 2.0 were used for sequencing.
Two high-quality DNA extracts, designated Pinermis_Ant (Antioquia) and Pinermis_Cor (Córdoba), were selected for shallow whole-genome sequencing (~7× coverage). Paired-end libraries (2 × 150 bp; ~350 bp insert size) were prepared using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs, USA; Cat. No. E7645L) following the manufacturer’s protocol, including size selection with AMPure XP magnetic beads (Beckman Coulter, USA; Cat. No. A63881). Library concentrations were measured using a Qubit 4 Fluorometer (Invitrogen, USA; Cat. No. Q33226) and the Qubit dsDNA HS Assay Kit (Cat. No. Q32854). Libraries were pooled equimolarly and sequenced on an Illumina NovaSeq X platform (Illumina, USA) at Macrogen Inc. (Seoul, South Korea), generating approximately 8–10 Gb of raw paired-end sequencing data per sample, corresponding to an estimated genomic coverage of ~7×.
The COI barcode region (cox1) was amplified using standard insect primers LCO1490 and HCO2198.15 PCR amplifications were carried out in 50 μL reactions containing 4 μL of genomic DNA template, 0.25 U μL−1 of Taq DNA Polymerase (Thermo Fisher Scientific, USA; Cat. No. EP0402), 5 μL of 10× PCR reaction buffer (supplied with the enzyme), 1 μL of 10 mM dNTP mix (Invitrogen, USA; Cat. No. 18427-013), and 1.5 μL each of forward and reverse primers (10 μM), with the remaining volume adjusted to 50 μL using nuclease-free water (Thermo Fisher Scientific, Cat. No. AM9937). Thermal cycling was performed in a T100 Bio-Rad 96-Well Thermal Cycler (Bio-Rad Laboratories, Inc., USA) under the following conditions: initial denaturation at 95 °C for 5 min; 45 cycles of denaturation at 95 °C for 40 s, primer annealing at 51 °C for 60 s, and extension at 72 °C for 45 s; followed by a final elongation step at 72 °C for 10 min. PCR products were purified and sequenced by Macrogen Inc. (Seoul, Korea).
A total of 20 COI sequences from P. inermis were analyzed together with 24 additional COI sequences from American Cryptorhynchinae species retrieved from GenBank. Sequence alignment was conducted in MAFFT v7.525,16 and pairwise genetic distances were calculated under the Kimura 2-Parameter (K2P) model using the ape v5.817 package in R. Intraspecific K2P distances were estimated only for species with more than four available COI sequences, providing an approximate measure of within-species variation in the barcode region. A Maximum likelihood (ML) phylogenetic tree based on COI sequences was conducted using the extended model selection followed by tree inference and ultra-fast non-parametric bootstrap with 1,000 replicates to evaluate node support in IQ-Tree 2.0.3.18
Raw Illumina paired-end reads were quality-filtered and trimmed using fastp,19 and the resulting high-quality reads were assembled de novo with SPAdes.20 We evaluated the completeness of the assembly using Benchmarking Universal Single Copy Orthologs (BUSCO v. 6.0)21 for both sequenced individuals (Antioquia and Córdoba samples) against endopterygota_odb12 database. Complete single-copy genes were extracted from both P. inermis (Antioquia and Córdoba) samples and annotated by Clusters of Orthologous Genes (COG) using eggNOG-mapper (http://eggnog-mapper.embl.de/).
Assembled contigs were screened for mitochondrial sequences by BLASTN comparison to reference mitogenomes from Cryptorhynchinae (Eucryptorhynchus brandti - NC_025945.1; E. chinensis - NC_026719.1; Trigonopterus selaruensis - NC_050886.1; T. tanimbarensis - NC_050887.1; T. jasminae - NC_050888.1; T. triradiatus - NC_050889.1; T. singkawangensis - NC_050890.1; T. carinirostris - NC_050891.1; T. kotamobagensis - NC_050892.1; T. porg - NC_050893.1). Mitochondrial genomes annotation was performed using GeSeq and OGDRAW webserver.22 The mitogenome organization was compared using the GLOBAL multi-GFF3 output retrieved from the OGDRAW webserver,22 and subsequently processed with a custom R script.
We obtained a total of 23.5 and 15.6 million reads for the P. inermis individuals from Antioquia and Córdoba, respectively ( Table 1). The assemblies yielded N50 values of 21 and 50, with approximately 213,000 and 233,000 contigs for the Antioquia and Córdoba individuals, respectively ( Table 1).
BUSCO analysis of the shallow-genome assemblies recovered, out of 3,754 expected Endopterygota genes, 52% complete and 30% fragmented in Pinermis_Ant, and 55% complete and 28% fragmented in Pinermis_Cor ( Figure 1a), yielding >45% non-complete (fragmented + missing) loci in both assemblies, consistent with notable fragmentation. Despite this, we identified 196 single-copy orthologs having non-stop codons shared by both samples that were assigned to 22 COG functional categories (Supplementary S1). The distribution was dominated by Function unknown (24.1%), followed by Translation, ribosomal structure and biogenesis (14.6%) and Transcription (8.5%) ( Figure 1b). Core cellular and metabolic processes were moderately represented, including Energy production and conversion (6.5%), Coenzyme transport and metabolism (5.5%), Amino acid transport and metabolism (5.0%), Intracellular trafficking, secretion, and vesicular transport (5.0%), and RNA processing; lipid metabolism; post-translational modification/chaperones; signal transduction each at ~4.5%. Carbohydrate metabolism reached 3.5%, whereas nucleotide metabolism and replication/repair were ~2.0% ( Figure 1b). Overall, even with fragmented assemblies, conserved informational functions are well captured, while a substantial fraction of single-copy orthologs remains uncharacterized.
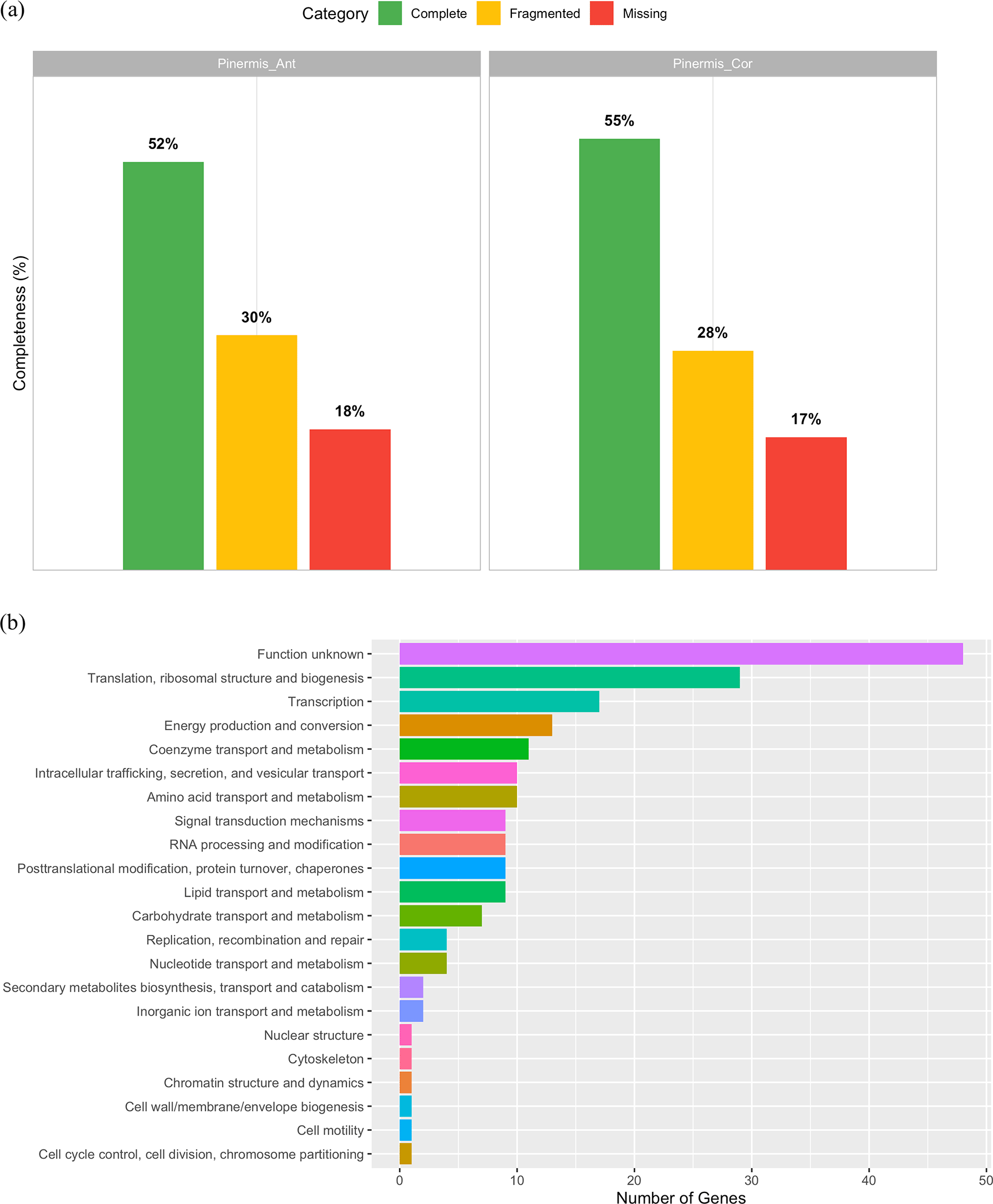
(a) BUSCO completeness profiles for two genome assemblies (Endopterygota_odb12; 3,754 genes): Pinermis_Ant (52% complete, 30% fragmented) and Pinermis_Cor (55% complete, 28% fragmented). (b) Distribution of COG functional categories for the 196 single-copy BUSCO orthologs recovered in both assemblies.
The representative mitochondrial genome of P. inermis was 15,120 bp in length and contained 36 features typically found in insect mitogenomes: 13 protein-coding genes (PCGs), two ribosomal RNAs (rRNAs), and 21 transfer RNAs (tRNAs). The only gene not identified was tRNA-Ile (trnI) ( Figure 2a). The overall nucleotide composition was strongly biased toward adenine and thymine (A+T = 77.02%), a pattern consistent with another weevil mitogenomes ( Figure 2b).
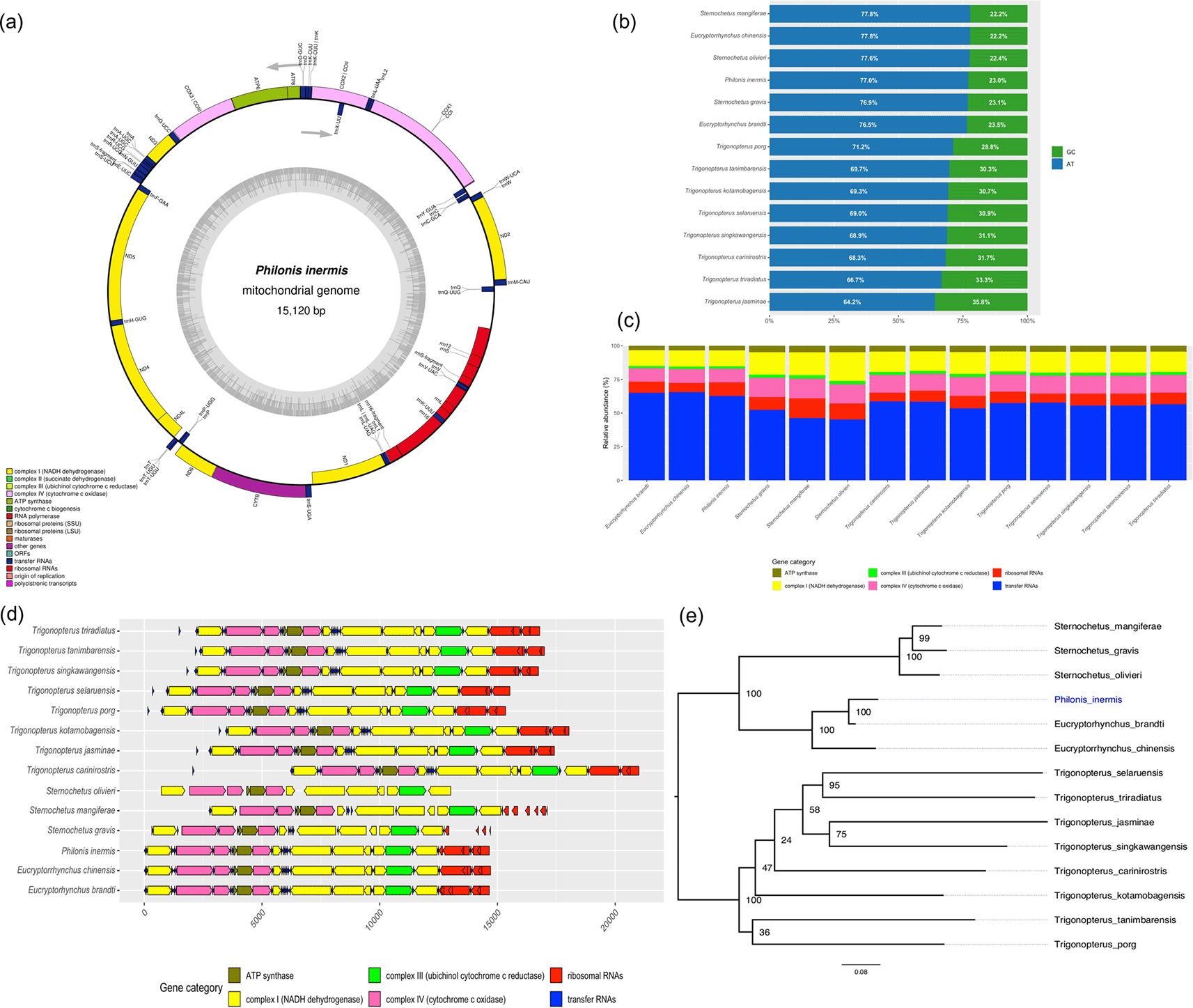
(a) Circular map of the complete mitochondrial genome of P. inermis showing gene annotation and organization. (b) Comparative nucleotide composition (AT vs. GC content) of P. inermis and other Cryptorhynchinae mitogenomes. (c) Relative abundance of annotated gene categories across P. inermis and related Cryptorhynchinae species. (d) Synteny and structural arrangement of annotated genes in P. inermis compared with related Cryptorhynchinae mitogenomes. (e) Maximum likelihood phylogenetic tree inferred from complete mitogenome sequences of P. inermis and representative Cryptorhynchinae species.
Comparative analyses revealed that Eucryptorhynchus spp. and P. inermis possess the highest total gene counts among the examined Cryptorhynchinae, primarily due to an increased number of tRNAs ( Figure 2c). In contrast, Sternochetus spp. exhibit a lower overall gene count, reflecting a reduction in tRNA genes. The numbers of protein-coding genes, rRNAs, and other categories remain largely conserved across genera, with only minor differences observed ( Figure 2c). Furthremore the mitochondrial genome structure of P. inermis was highly conserved and syntenic with other Cryptorhynchinae species, without major rearrangements, despite minor variations in gene spacing and orientation ( Figure 2d). These patterns indicate strong conservation of mitochondrial gene content and organization within Cryptorhynchinae, with variation mainly associated with tRNA gene numbers. The preliminary phylogenetic reconstruction based on complete mitochondrial genome sequences placed P. inermis as sister to the Eucryptorrhynchus clade with strong support (BS = 100), while Sternochetus species clustered into a distinct lineage within Cryptorhynchinae. In contrast, relationships among Trigonopterus species were less resolved, with several nodes showing low support ( Figure 2e). These results should be considered preliminary, as the current topology is influenced by the limited and uneven mitogenomic representation available in public databases.
The COI barcode sequences of P. inermis from multiple Colombian populations exhibited extremely low intraspecific divergence, with pairwise Kimura 2-Parameter (K2P) distances ranging from 0 to 0.006 ( Figure 3a). This low genetic variability, evident from both the distance matrix and density distributions, supports the genetic homogeneity of P. inermis across sampled localities, especially when compared with other Cryptorhynchinae species ( Figure 3b). Comparison with additional Cryptorhynchinae COI sequences from GenBank revealed a clear barcode gap: intraspecific K2P distances in P. inermis were substantially lower than interspecific distances across Cryptorhynchinae, which were typically greater than 0.15 ( Figure 3c). This distinct separation highlights the effectiveness of COI barcoding for reliably identifying P. inermis and distinguishing it from related taxa. A maximum likelihood tree constructed from COI sequences ( Figure 3d) clustered all P. inermis specimens together with strong bootstrap support, forming a well-defined lineage among Neotropical Cryptorhynchinae and further validating its molecular distinctiveness.
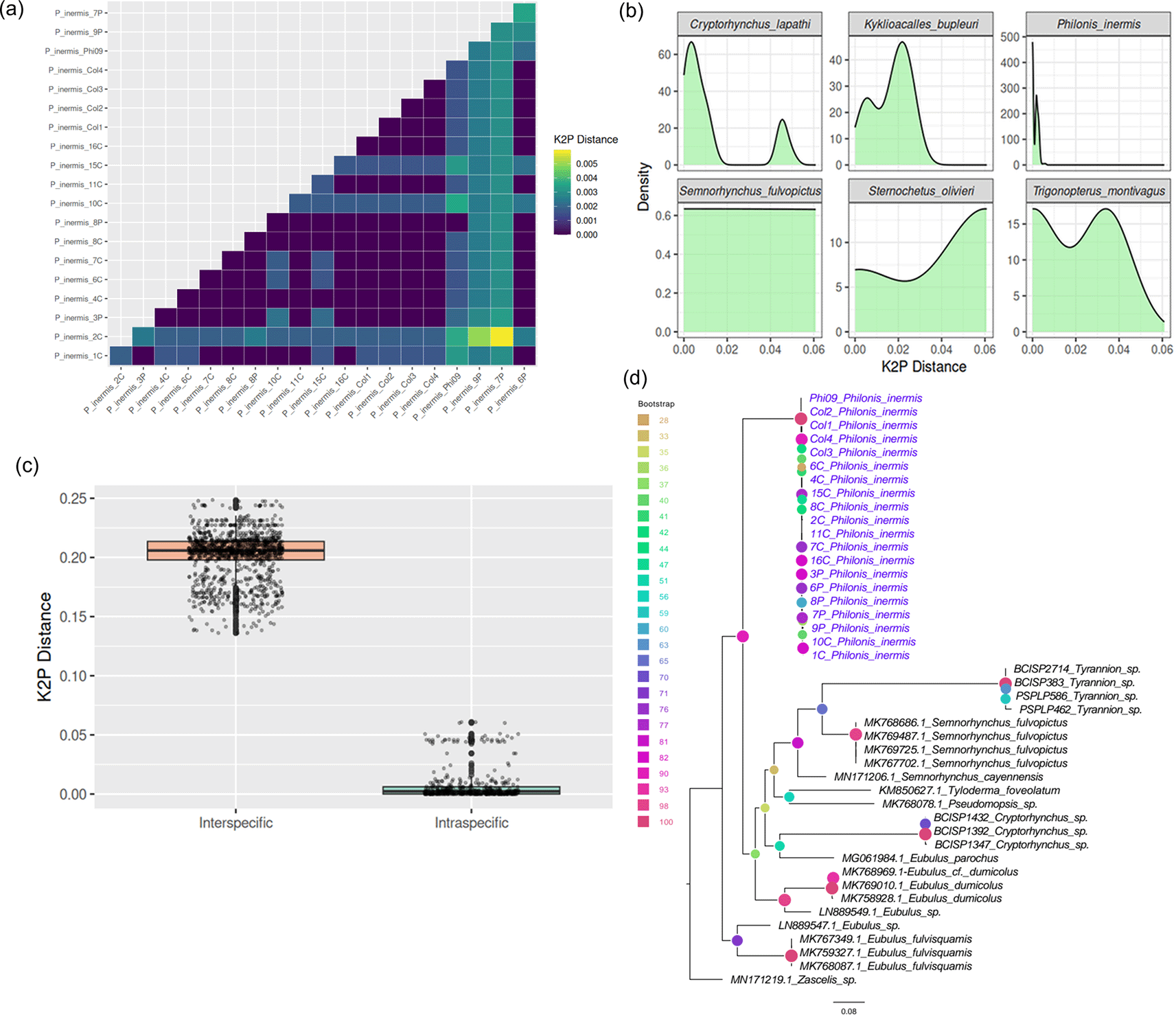
(a) Pairwise Kimura 2-Parameter (K2P) distances among Colombian P. inermis specimens. (b) Density plots of K2P distances in P. inermis compared with selected Cryptorhynchinae species. (c) Comparison of intra- and interspecific K2P distances across Cryptorhynchinae, illustrating a clear barcode gap. (d) Maximum likelihood tree based on COI sequences showing P. inermis as a distinct, well-supported clade among Neotropical Cryptorhynchinae.
Despite family Curculionoidae is rendering as one of the most diverse groups of Coleoptera, encompasses approximately 62,000 species distributed among 5,800 described genera,23 many attributes about genome structure, diversity, and ecological role of Neotropical species belonging to Crypthorynchinae subfamily such P. inermis is poorly unknowledge so far.
The weevils of the Neotropical genus Philonis (Curculionidae: Cryptorhynchinae) represent a largely unexplored lineage within the diverse assemblage of gall-inducing insects, here we conducted a pioneer information about shadow-genome, mitogenome and DNA barcode accuracy in order to further explores the genetic diversity and structure to future evolutionary and population-genetic studies besides applied in biological-control fields for invasive vine P. foetida L.
Although BUSCO profiles indicate that neither assembly captures the full gene space—consistent with the limited depth typical of genome skimming—we nonetheless recovered 196 BUSCO single-copy orthologs shared by both individuals, 191 of which contain intact ORFs and could be assigned to 22 COG functional categories. This set captures core informational functions and provides a robust marker panel for comparative and genome-scale analyses in P. inermis and related Neotropical Cryptorhynchinae, while the sizable proportion of unclassified COGs highlights persistent knowledge gaps in this lineage. Future work will integrate long-read sequencing and Hi-C scaffolding to resolve repetitive and complex regions, reduce fragmentation, and achieve chromosome-scale assemblies for both individuals.
The mitochondrial genome of P. inermis exhibits nucleotide a composition (A+T), gene order and orientation that are highly conserved, matching the ancestral insect mitogenome architecture. Comparative analyses with complete mitogenomes available for other Cryptorhynchinae genera revealed structural and synteny conservation across the group.6,7,9,24 Similar as reported for other Asian species such Eucryptorrhynchus chinensis and E. brandti the mitogenome map of P. inermis deficiency of tRNA-Ile gene.7 The trnI gene is situated within the variable control region, where high sequence divergence confounds automated annotation and precise boundary determination, making its detection a common challenge.25
Mitogenome-based tree place Philonis close to Eucryptorrhynchus, but we treat this as preliminary, given the limited and uneven mitogenomic sampling across Cryptorhynchinae. Broad, multi-gene frameworks show that relationships above the genus level can be difficult to stabilize in this subfamily, and that extensive sampling across loci is required to resolve deeper nodes.26,27 In particular, Riedel et al.27 recovered a large-scale molecular phylogeny that points to an American origin for Cryptorhynchinae and highlights the value of integrating mitochondrial and nuclear markers for robust placement. Our results align with this perspective: the topology we report is a useful working hypothesis that should be tested with expanded taxon and locus coverage.
If proximity to Eucryptorrhynchus is confirmed, the comparison is ecologically instructive. Ailanthus altissima (Simaroubaceae), the tree-of-heaven, is a fast-growing invasive tree native to China that readily colonizes disturbed habitats and can displace native vegetation. Within this host context, E. scrobiculatus and E. brandti are highly specialized on A. altissima. In parts of their native range, they are considered pests; however, they have also been evaluated as potential biological control agents where A. altissima is invasive, and climate-suitability assessments suggest differential responses of the two congeners to future climates.7,28 This dual “pest vs. biocontrol” context underscores why clear systematics and robust diagnostics matter when considering host-specific herbivores for applied programs.
Interestingly, all other cryptorhynchine species with complete mitogenomes currently reported so far are documented agricultural pests of economic importance in Asia or Oceania, remaining significative gap for Neotropical species. In contrast, P. inermis is a stem-galling specialist with a narrow host range restricted to P. foetida, an invasive vine in Australia.29 This high degree of host specificity, combined with its ecological role, highlights its potential as a classical biological control agent, distinguishing it from other pest-associated weevils (Clavijo-Giraldo et al., 2025. Unpublished).
At a broader scale, historical biogeography indicates a complex macroevolutionary backdrop for Cryptorhynchinae, with major diversity centers in the Neotropics and Australasia and signals consistent with an American origin.3,27,30 Comparative insights from flightless Trigonopterus show repeated crossings of major biogeographic barriers (e.g., Wallace’s Line), rapid radiations and finely structured endemism, and these features complicate deep-time reconstructions when sampling is sparse.31 These patterns provide a cautionary frame for interpreting single-marker or low-taxon trees and reinforce the need for expanded, balanced sampling when refining the placement of Philonis.
Assessment for COI gene DNA barcoding regions indicates high accuracy on molecular identification of P. inermis, with minimal genetic divergence among Colombian individuals from three populations (K2P distances ≤ 0.006). This low intraspecific variability underscores the genetic coherence of the species and confirms that the sampled populations across Colombia belong to the same taxonomic unit. Furthermore, both inter – and intraspecies genetic distance gap support the barcode as suitable tool in the Neotropical cryptorhynchine species, further than reported for other Asian, European and Oceanian species.31,32 Under this scenario, our COI results supports unambiguous diagnosis of P. inermis. This pattern aligns with the central rationale for DNA barcoding as a standardized, taxonomically integrative tool.12,33 The maximum likelihood tree constructed from COI sequences clustered all P. inermis specimens together with strong statistical support, further confirming its genetic uniformity and separation from other Neotropical taxa. Collectively, these barcode results provide a robust molecular baseline for species identification and support the consideration of P. inermis as a promising candidate for biological control programs targeting P. foetida.
From an applied perspective, the combination the mitogenome and COI barcoding supports accurate recognition across Colombian populations, which is a prerequisite for any subsequent risk assessment. Furthermore, this approach strengthens integrative taxonomy, biosecurity, and classical weed biocontrol workflows. Barcodes offer interoperable, scalable diagnostics for look-alike taxa and immature stages, facilitate data sharing across laboratories and jurisdictions, and support post-release monitoring, both of which are key steps to minimize non-target risk.12,33,34 The global invasive potential documented for S. mangiferae under climate change further illustrates why strong diagnostics and early detection pipelines are increasingly critical for curculionid lineages with agricultural relevance.35
Limitations and next steps follow directly from our results. First, phylogenetic inferences from current mitogenome sampling should be treated as provisional. Resolving deeper nodes will require denser Neotropical sampling, including close relatives of Philonis, and multi-locus or genomic matrices that integrate nuclear markers. Second, expanding the geographic and host-associated sampling for COI (and additional markers) will help quantify population structure and confirm the breadth of the barcode gap. Third, low-coverage, short-read genome skimming inherently underrepresents repetitive and GC-extreme regions and can collapse recent paralogs, yielding fragmented assemblies and biasing functional annotations toward conserved single-copy genes; increasing sequencing depth and integrating long reads and Hi-C will mitigate these biases. Together, these steps will strengthen both the systematic placement of Philonis and the applied utility of its genetic resources for monitoring and potential biocontrol assessment.
We provide the first mitogenomic reference for P. inermis (15,120 bp; ~77% A+T), with conserved gene order and an undetected trnI likely obscured within the variable control region. Maximum-likelihood analyses recover P. inermis as a well-supported clade and tentatively near Eucryptorrhynchus, a hypothesis that awaits denser taxon and locus sampling. COI barcodes from 20 Colombian individuals show extremely low intraspecific divergence (K2P ≤ 0.006) and a pronounced barcode gap from other American cryptorhynchines, enabling reliable, field-ready diagnostics. Shallow genome sequencing recovered 196 single-copy orthologs, furnishing anchors for future genome-scale phylogenetics and comparative genomics. Together, these resources validate COI barcoding for accurate identification, establish a molecular foundation for systematics and population studies, and inform risk-aware evaluation of P. inermis as a host-specific candidate for classical biological control of P. foetida. Priority next steps include long-read and Hi-C assemblies and expanded geographic and taxonomic sampling—including nuclear markers—to stabilize deeper relationships and quantify population structure.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
All reviewer comments have now been thoroughly addressed, and the manuscript has been revised accordingly. In particular, we have (i) performed and documented a contamination analysis based on taxonomic classification of unmapped reads, (ii) expanded the Discussion to better contextualize the use and limitations of mitochondrial genomes in phylogenetic inference, (iii) clarified methodological terminology and analytical scope, and (iv) refined several sections to improve clarity and consistency. We believe these revisions have strengthened the methodological rigor and interpretative framework of the study, and we are grateful to the reviewer for their insightful suggestions, which substantially improved the quality of the manuscript.
All reviewer comments have now been thoroughly addressed, and the manuscript has been revised accordingly. In particular, we have (i) performed and documented a contamination analysis based on taxonomic classification of unmapped reads, (ii) expanded the Discussion to better contextualize the use and limitations of mitochondrial genomes in phylogenetic inference, (iii) clarified methodological terminology and analytical scope, and (iv) refined several sections to improve clarity and consistency. We believe these revisions have strengthened the methodological rigor and interpretative framework of the study, and we are grateful to the reviewer for their insightful suggestions, which substantially improved the quality of the manuscript.