Keywords
Macroderma, Megadermatidae, threatened species, arid zone, Australia, Chiroptera
This article is included in the Genomics and Genetics gateway.
Macroderma, Megadermatidae, threatened species, arid zone, Australia, Chiroptera
The sole member of the genus Macroderma, the ghost bat (M. gigas) is endemic to Australia and is the world’s largest microbat (wingspan of 64 – 72 cm; Churchill 2008). It is the only extant representative of the Family Megadermatidae within Australasia, with its closest relatives including the greater false vampire bat (Lyroderma lyra), lesser false vampire-bat (Megaderma spasma) and Thongaree’s disc-nosed bat (Eudiscoderma thongareeae) found in Asia, and the heart-nosed bat (Cardioderma cor) and yellow-winged bat (Lavia frons) found in Africa. Within Australia, the ghost bat has a current distribution spanning multiple disjunct populations across the northern half of the continent, having contracted from scattered populations through central and southern Australia during the Holocene due to increasing aridification ( Figure 1A; Molnar et al. 1984). Some central Australian populations were estimated to have been extant up to about 50 to 70 years ago (Churchill & Helman 1990). The species is highly distinctive, exhibiting large ears with a forked tragus joined by a bridge of skin above the head, large eyes, a long, simple nose-leaf and light grey to light brown fur with a paler abdomen increasing in lightness in inland populations ( Figure 1B; Churchill 2008). The ghost bat is also Australia’s only obligate carnivorous bat, known to prey on invertebrates, birds, reptiles, amphibians, and small mammals, including other bats (Tidemann et al. 1985; Diete et al. 2016; Arteaga Claramunt et al. 2018; Start et al. 2019). A common hunting strategy involves the bat observing prey from a vantage point before ambushing and capturing the prey item, then carrying it to feeding perches for consumption (Diete et al. 2016). They capture much of their prey by gleaning from the ground, and also from foliage and branches as is evident from the many species of small vertebrate and flightless insects recorded from accumulations of prey remains and stomachs (Vestjens & Hall 1977; Tidemann et al. 1985; Schulz 1986). They are also known to hawk insects and small bats from the air (Kulzer et al. 1984; Pettigrew et al. 1986).
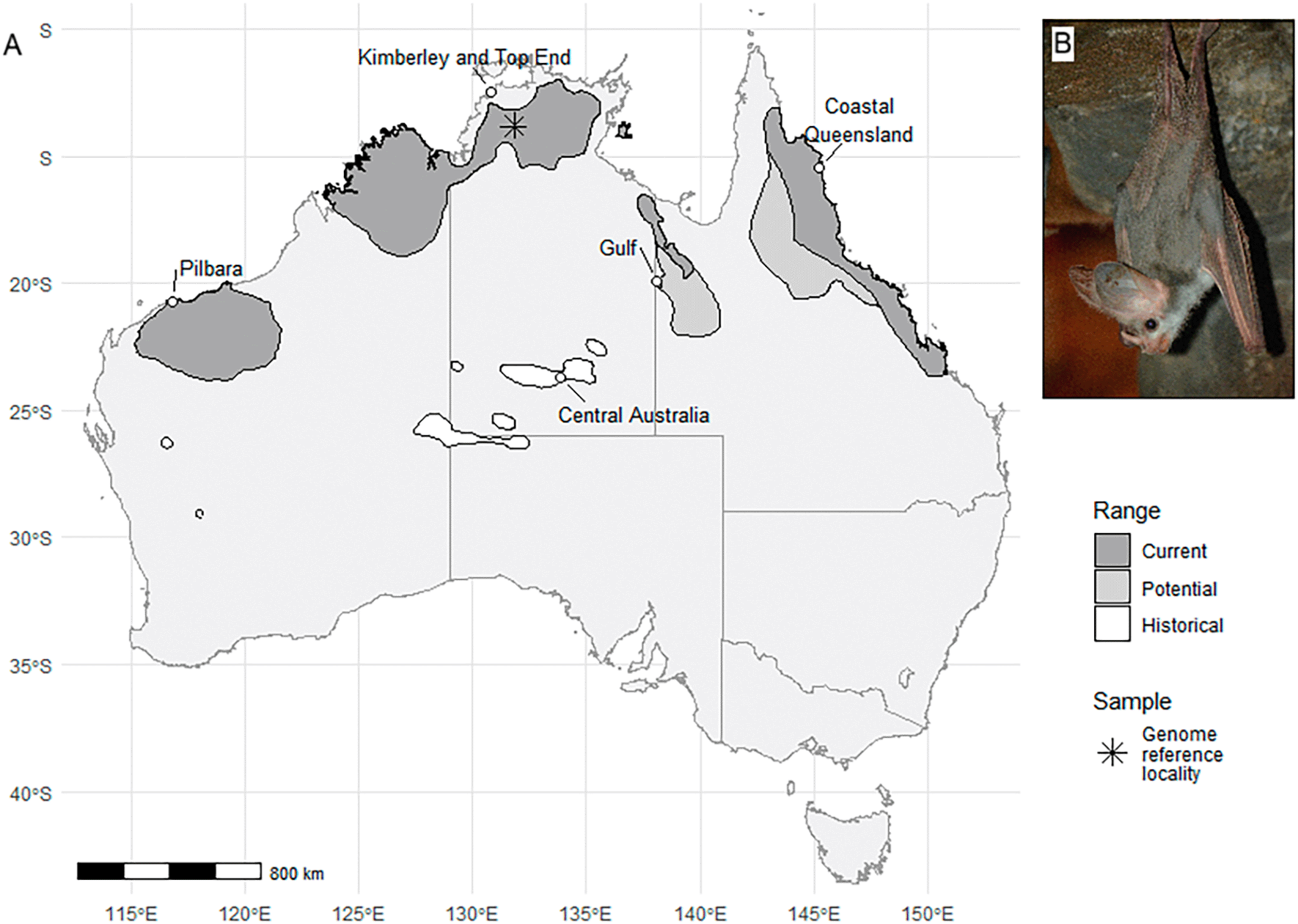
Due to pervasive threats and ongoing declines, the ghost bat is currently listed as Vulnerable by the IUCN Red List and under the Australian Commonwealth Environmental Protection and Biodiversity Conservation Act 1999 (EPBC Act; up-listed from Near Threatened in 2016; Threatened Species Scientific Committee 2016). Whilst the ghost bat’s distribution spans diverse habitats from the arid western Pilbara population to tropical savanna woodlands and rainforests across northern Australia ( Figure 1A), it is constrained by the availability of roosting habitat with a suitable microclimate. Roost sites occupied year-round by M. gigas are generally deep natural caves or disused mines with a stable microclimate, with temperatures 23 – 28°C and moderate to high (50 – 90%) relative humidity, and the ceiling at least 2 m from the floor (Churchill & Helman 1990). In many regions, ghost bat roosting habitat coincides with mining activity, with bat colonies threatened directly by the removal or damage of caves and historical underground mines through mineral extraction, and with habitat quality affected indirectly by dust, noise and artificial lighting associated with mining operations (Threatened Species Scientific Committee 2016; Armstrong et al. 2021; Cramer et al. 2022). In addition, many disused mines that currently support ghost bat colonies are at risk of collapse or disturbance (Armstrong & Anstee 2000). Degradation of foraging habitat through livestock grazing, inappropriate fire regimes and weed encroachment can impact the viability of ghost bat colonies, and bats are at risk of entanglement in barbed wire stock fencing when flying low to the ground during foraging bouts (Armstrong & Anstee 2000; Threatened Species Scientific Committee 2016; Bradley et al. 2023).
Genetic studies of the ghost bat have been limited but suggest that, across their disjunct distribution, ghost bats exhibit strong regional genetic structuring with little evidence of broad-scale gene flow, consistent with a pattern of long-term female philopatry (Worthington Wilmer et al. 1994; Worthington Wilmer et al. 1999). At a finer-scale, a within-region genetic study of ghost bats in the arid Pilbara also found support for a pattern of female philopatry with females showing more restricted dispersal, both at landscape and finer-scales (up to ~30 km) (Umbrello et al. 2025). The lack of migration between colonies, especially of females, is an additional threat to species’ long-term viability as there is a low probability of re-establishing colonies following localised extirpation (Threatened Species Scientific Committee 2016; Augusteyn et al. 2017). Due to the species’ sensitivity to disturbance, a novel monitoring method has been established to identify individual bats for mark-recapture analysis based on genetic analysis of faecal samples (‘genetic tagging’), which is providing insights into spatial and temporal patterns of ghost bat roost use (Augusteyn et al. 2017; Ottewell et al. 2020; Thavornkanlapachai et al. 2024).
Bats account for ∼20% of all living mammals (c. 1,500 species) and are found across the globe, absent only from the extreme polar regions. Comparative genome analysis, primarily driven by genome sequencing efforts through the Bat1K initiative (Teeling et al. 2018), is revealing intriguing insights into the unique adaptations attributed to bat species, including positive selection on hearing-related genes in the ancestral branch of bats related to laryngeal echolocation, selection and loss of some immunity-related genes and expansions of anti-viral APOBEC3 genes, highlighting some molecular mechanisms that may contribute to the exceptional immunity of bats (Jebb et al. 2020) and positive selection of genes involved diet specialisation (Moreno Santillan et al. 2021; Blumer et al. 2022). Here we report on the complete genome sequence of the ghost bat, Macroderma gigas, of interest as the sole representative of the genus Macroderma and only species of the Megadermatidae to inhabit arid environments. We present the ghost bat genome to be 2.00 Gb with 313 scaffolds, the longest scaffold being 192.28 Mb. Our annotation shows there to be 48,477 genes (97.4% complete vertebrate BUSCO) and comparison with other bat genomes showed relatively conserved features across the Chiroptera.
A single male ghost bat (MG_PC43) was captured in a mist net near the Pine Creek township in the Northern Territory using the Squabble vocalisation acoustic lure protocol (Hanrahan et al. 2024). The bat was sedated using isoflurane administered via a custom-built chamber, and the sedated bat euthanised via cervical dislocation. Tissue samples were dissected from identifiable organs and flash frozen in liquid nitrogen before long-term storage at -80°C. High molecular weight (HMW) DNA was then extracted from liver and kidney tissue using the Nanobind Tissue Big DNA Kit v1.0 (Circulomics). Additional HWA DNA was extracted from kidney tissue using the Nanobind Tissue Big DNA Kit v1.0 (Circulomics). A Qubit fluorometer was used to assess the concentration of DNA with the Qubit dsDNA BR assay kit (Thermo Fisher Scientific). Total RNA was extracted from liver, heart, brain, spleen and testis using the RNeasy Plus Mini Kit (Qiagen) with DNAse treatment (Qiagen). RNA quality was determined using the NanoDrop (Thermo Fisher Scientific) and RNA integrity (RIN) score determined using the Bioanalyzer RNA nano 6000 kit (Agilent 2100).
Pooled kidney and liver HMW DNA was sent for Pacific Biosciences High Fidelity (PacBio HiFi) library preparation with the SMRTbell Express Template Prep Kit 2.0 (Pacific Biosciences) and sequencing on two single molecule real-time (SMRT) cells of the PacBio Sequel II at the Australian Genome Research Facility (St Lucia, Australia). The resulting data showed lower quality and reduced read length to what was expected so an additional two SMRT cells were used to sequence HMW extracted solely from kidney tissue. Heart tissue for the was sent for HiC Armina 2.0 library preparation and sequenced on NovaseqX at Biomolecular Resource Facility (Australian National University). Total RNA from the liver, heart, brain, spleen and testis was sequenced as 100 bp paired-end (PE) reads using an Illumina Novaseq 6000 with Illumina Stranded mRNA library preparation at the Ramaciotti Centre for Genomics (University of New South Wales, Kensington, Australia).
To generate the reference genome for the ghost bat we used the VGP assembly pipeline on the Galaxy Australia interface (Batut et al. 2018; The Galaxy Community 2022; Hiltemann et al. 2023; Lariviere et al. 2024). Briefly, HiFi reads were quality trimmed and reads containing adapters removed using Cutadapt v4.6 (RRID:SCR_011841; Martin 2011), genome size and k-mers were estimated using Meryl v1.3 (RRID:SCR_026366; Rhie et al. 2020) and GenomeScope v2.0 (RRID:SCR_017014; Ranallo-Benavidez et al. 2020). Genome assembly was performed using hifiasm v0.19.8 (RRID:SCR_021069) in the Hi-C phased mode (Cheng et al. 2021) using HiFi reads to assembly contigs and Hi-C reads to identify haplotypes, with the assembly quality assessed using gfastats v1.3.6 (RRID:SCR_026368; Formenti et al. 2022) and BUSCO v5.5.0 (RRID:SCR_015008) with both vertebrata_odb10 (n=3354) and mammalia_odb10 (n = 9226) lineages (Simao et al. 2015). Genome completeness and base accuracy was also determined Merqury v1.3 (RRID:SCR_022964; Rhie et al. 2020). Hi-C reads were then used to scaffold contigs of the primary assembly, scaffolding was performed using YaHS v1.2a.2 (RRID:SCR_022965; Zhou et al. 2022) with contact maps generated with PretextMap v0.1.9 (RRID:SCR_022024). The scaffolded assembly was then manually curated in Juicebox v2.17.00 (RRID:SCR_021172; Durand et al. 2016). Repetitive elements of the genome were identified, classified and masked using RepeatModeler v2.0.4 (RRID:SCR_015027) and RepeatMasker v4.1.5 (RRID:SCR_012954; Flynn et al. 2020).
We investigated synteny between the ghost bat genome and six other bat species (Rhinolophus ferrumequinum, Rousettus aegyptiacus, Phyllostomus discolor, Myotis myotis, Pipistrellus kuhlii and Molossus molossus) generated as part of the Bat1K project (Jebb et al. 2020). We used the DEEPSPACE v0.1 (https://github.com/jtlovell/DEEPSPACE) on scaffolds larger than 20Mb to identify regions of synteny across all seven species in R v4.3.1 (RRID:SCR_001905; R Core Team 2025).
The mitochondrial genome was identified from the reference genome assembly using MitoHiFi v1.0.1 (RRID:SCR_026369; Allio et al. 2020; Uliano-Silva et al. 2023) and visualised using Proksee (Grant et al. 2023). MitoHiFi identified a mitochondrial genome for the ghost bat on NCBI (NC_057635.1).
Transcriptome assembly was performed on the University of Sydney High Performance Computer, Artemis, following standard methods outlined in Silver et al. (2024). Raw transcriptome reads were quality assessed pre and post trimming with FastQC v0.11.8 (RRID:SCR_014583; Andrews 2010). Trimmomatic v0.39 (RRID:SCR 011848; Bolger et al. 2014) with the parameters SLIDINGWINDOW:4:5, LEADING:5, TRAILING:5 and MINLEN:25 and ILLUMINACLIP:2:30:10 with the TruSeq3-PE adapters was used to quality trim reads. The repeat masked genome was indexed and trimmed reads aligned using the -dta parameter with hisat2 v2.1.0 (RRID:SCR_015530; Kim et al. 2019). Resulting sam files were converted to bam format and sorted using samtools v1.9 (RRID:SCR_002105; Danecek et al. 2021). Stringtie v2.1.6 (RRID:SCR_016323; Pertea et al. 2015) was used to generate a GTF for each transcriptome. Stringtie v2.1.6 with the -merge parameter merged transcripts into a global transcriptome retaining only transcripts with an FPKM > 0.1 and length > 30. CPC2 v2019-11-19 (Kang et al. 2017) was used to predict coding potential, and only transcripts predicted to be coding were retained. Transdecoder v2.0.1 (RRID:SCR_017647; Haas 2022) was used to predict open reading frames in the global transcriptome with a minimum transcript length of 20. Transcriptome completeness was assessed using BUSCO v5.5.0 with the vertebrata_odb10 (n= 3354) and mammalia_odb10 (n = 9226) lineage on Galaxy Australia.
Genome annotation was performed using FGENESH++ v7.2.2 (Softberry, RRID:SCR_018928; Solovyev et al. 2006) using the longest open reading frame as predicted from the global transcriptome, non-mammalian settings and optimised parameters supplied with the big brown bat (Eptesicus fuscus) gene finding matrix. BUSCO v5.5.0 in protein mode was run on Galaxy Australia to assess the completeness of the annotation with the vertebrata_odb10 (n = 3354) and mammalia_odb10 (n = 9226) lineage.
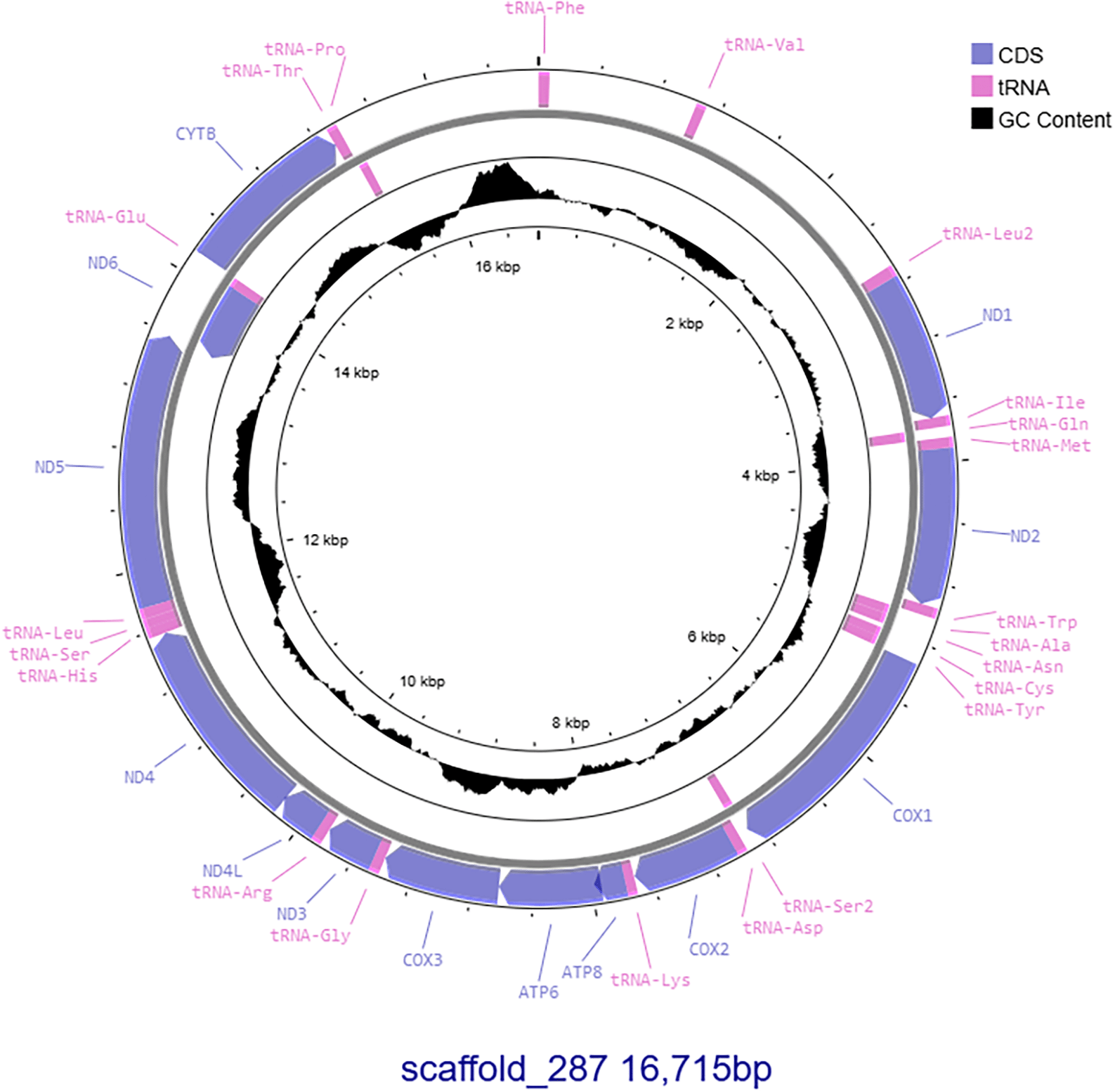
PacBio HiFi sequencing of DNA pooled from liver and kidney tissue resulted in 8.16 Gb of reads with further sequencing of DNA extracted from kidney resulted in an additional 47.17 Gb of data for a total of 55.33 Gb of HiFi reads or approximately 27.5x coverage (assuming a 2Gb genome size). The hifiasm assembly of the ghost bat genome using PacBio HiFi and Arima HiC data generated a primary scaffolded assembly 2.00 Gb in size containing 313 scaffolds. The longest scaffold is 192.28 Mb in length with scaffold N50 and L50 100.01 and 19, respectively ( Table 1). An alternate haplotype assembly was also generated, which is 1.90 Gb in size and 407 contigs ( Table 1). BUSCO analysis indicated both haplotypes are highly complete assemblies with 96.3% and 93.8% of complete mammalian BUSCOs present in the primary and alternate haplotypes, respectively. Merqury analysis also indicated both assemblies are highly complete, with QV of both assemblies >69 and K-mer completeness of the primary and alternate assembly 92.91% and 88.24%, respectively, with both assemblies containing 99.64% of all K-mers sequenced. Repeat masking identified a total of 29.09% of the genome as repetitive elements, with the majority of these identified as Long Interspersed Nuclear Elements (LINEs; 15.25%; Table 2), similar to other bat species (Driller et al. 2024). MitoHiFi identified scaffold_287 as the complete mitochondrial genome, which is 16,715 bp and contains 13 genes and 22 tRNAs ( Figure 2). Comparison of the ghost bat to other bat genomes showed relatively conserved features across the Chiroptera ( Figure 3).

Sequencing of mRNA resulted in between 88.8M (spleen) and 132.6M (liver) reads and after trimming, greater than 99.9% of reads were retained. All individual tissues had alignment rates of greater than 96% (liver: 98.08%, heart: 98.16%, brain: 97.60%, testis: 97.33%, and spleen: 96.16%) against the repeat masked genome. A total of 96,474 transcripts were predicted to have coding potential, with 22,883 longest open reading frame transcripts used as mRNA evidence for input into genome annotation with Fgenesh++. BUSCO suggested a highly complete transcriptome with 92.8% of complete mammalian BUSCOs present ( Table 3). Genome annotation predicted a total of 48,477 protein-coding genes with BUSCO suggesting a complete annotation with 77.2% of complete mammalian BUSCOs present.
Tissue samples were obtained under Ethics Permit A20007, issued from Charles Darwin University on 23 September 2020.
1. BioPlatforms Australia data portal: The Threatened Species Initiative https://data.bioplatforms.com/organization/threatened-species
The project contains the following underlying data by Dataset ID:
102.100.100/419498: Ghost Bat, Reference Genome, Illumina-HiC, heart
102.100.100/358726: Ghost Bat, Reference Genome, Pacbio HiFi, Liver, heart and kidney
102.100.100/358738: Ghost bat, Transcriptomes, Illumina FASTQ, Spleen, Testis, Brain, Heart, Liver
2. NCBI: BioProject PRJNA1311142 Macroderma gigas isolate: MG_PC43 (Australian ghost bat) https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1311142
The project contains the following underlying data by Accession Number:
SRR35162029 MG_PC43_spleen_mRNA
SRR35162030 MG_PC43_testes_mRNA
SRR35162031 MG_PC43_brain_mRNA
SRR35162032 MG_PC43_heart_mRNA
SRR35162033 MG_PC43_liver_mRNA
SRR35162034 MG_PC43_liver_mRNA
SRR35162035 MG_PC43_pooled_liver_heart_kidney_HiFi
SRR35162036 MG_PC43_kidney_HiFi
JBQVVY000000000 Genome assembly Macroderma gigas
3. FigShare: Checklist for ARRIVE.pdf for “The complete genome sequence of the ghost bat, Macroderma gigas” is available at https://doi.org/10.6084/m9.figshare.30736502 (Silver, Luke et al., 2025).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)