Keywords
lncRNA, RNA pull-down, RNA-RNA interactions
This article is included in the Bioinformatics gateway.
lncRNA, RNA pull-down, RNA-RNA interactions
Despite their abundance in the human transcriptome — rivalling protein-coding genes in number1 — the functional characterisation of long non-coding RNAs (lncRNAs) has been complicated by their low expression levels, tissue-specificity,2 and inconsistent evolutionary signatures.3 Nonetheless, conserved functional characteristics, such as genomic synteny and RNA structure,4 indicate essential biological functions. A defining property of many lncRNAs is their nuclear localisation and direct engagement with chromatin, where they orchestrate epigenetic states and three-dimensional genome architecture,5–7 establishing them as fundamental components of gene regulatory networks.
CHASERR exemplifies this paradigm as a master regulator. It is transcribed adjacent to and exerts feedback control over CHD2, a chromodomain helicase DNA-binding protein essential for nucleosome remodelling.8 This positions CHASERR not merely as a target of the epigenetic machinery but as its supervisor. The discovery that haploinsufficiency of CHASERR underlies a severe genetic syndrome stands as a landmark confirmation of its essential role in human development and genomic integrity.9
The molecular basis for this profound regulatory capacity, however, remains opaque. While CHASERR is implicated in diseases from cancer to neurodegeneration,9–12 and computational models predict it forms multiple RNA-RNA interactions,13 a fundamental gap persists: there is no direct experimental evidence defining CHASERR’s native RNA interactome or its potential for direct DNA engagement. The precise physical partners through which it operates are unknown, and whether it functions via post-transcriptional sponging, transcriptional regulation, or both is undefined. This lack of a biochemically defined mechanism severely limits our mechanistic understanding of its cellular and disease roles.
Here, we bridge this gap by experimentally mapping the CHASERR interaction landscape. We combine RNA pull-down sequencing with promoter-binding analysis to demonstrate that CHASERR scaffolds a specific network of non-coding RNAs — including RMRP and CRNDE — while also binding directly to gene promoters. This dual functionality establishes CHASERR as a direct transcriptional and post-transcriptional hub, providing the physical and mechanistic foundation for its role as a master epigenetic regulator.
RNA pull-down was conducted in three replicates according to the protocol described in Desideri et al.14 To enrich the nuclear RNA fraction, cells were permeabilised for 15 minutes on ice in the 50 ul/cm2 of the following buffer: Triton-X100 0.5% (v/v), NaCl 150 mM, Tris pH 7.5 50 mM, MgCl2 3 mM, DTT 1 mM, Protease inhibitor cocktail 1x, RNAse inhibitor 0.2 U/ul. This buffer was carefully aspirated, and remaining nuclei were gently rinsed with the same buffer without Triton-X100 for 1 minute on ice. Primers used for CHASERR pull-down are listed in Supplementary Table 1.15
The reads for three replicates were mapped to the hg38 genome using STAR16 with the option clip5pNbases 0 3. The reads were counted in gene features from gencode v48 annotation using STAR quantMode and in promoter regions identified by FANTOM65,17 using Rsubread featureCounts v2.16.1.18 The features with 0 expression in the pull-down were excluded from the further analysis. For gene feature quantification, the fibroblast expression table was downloaded from FibroDB19,20 for background normalisation; samples with TGF-beta treatment were excluded. TPM normalisation was applied to each gene, and log10 values with pseudo count were used for further analysis. For promoter quantification, FANTOM6 expression for the control samples was used as a normalisation background; FANTOM6 expression and pull-down expression were converted to CPM, and log10 values with pseudo count were used for further analysis.
To assess the potential RNA-RNA interaction targets, metric A was calculated for each gene as the absolute deviation metric of pull-down normalised expression from CHASERR pull-down normalised expression.
Gene Ontology Biological Process enrichment analysis was performed using clusterProfiler v4.14.321 with a significance threshold of q-value < 0.05, genes with non-zero expression in pull-down were used as the universe set.
Differential expression analysis to compare the CHASERR knockdown and control states was performed at the level of individual promoter regions separately for the ASO_G0272888_AD_07 (ASO 07) and ASO_G0272888_AD_10 (ASO 10). The analysis was performed using DESeq2,22 and regions with an FDR < 0.05 and a |logFC|> 0.1 were selected for further analysis.
To define the CHASERR interactome, we performed RNA pull-down followed by sequencing in primary fibroblasts. This revealed a network highly enriched for non-coding RNAs. CHASERR itself accounted for 36% of genome-mapped reads, confirming specific enrichment.
Since highly abundant RNAs are more likely to be recovered non-specifically, we normalised pull-down expression to a fibroblast transcriptome baseline.19 This revealed a significant correlation between pull-down and baseline expression (Pearson r = 0.5033, p-value < 2.2e-16), establishing a general relationship, while CHASERR itself showed 5-fold specific enrichment (Figure 1A). To distinguish genuine interactors from this abundant background, we reasoned that true binding partners should be co-enriched to a level approximating CHASERR’s own abundance. We therefore selected RNAs whose pull-down abundance most closely matched that of CHASERR; the 1% of genes with the smallest deviation from CHASERR’s level were classified as high-confidence interactors (Figure 1B, Supplementary Table 215).
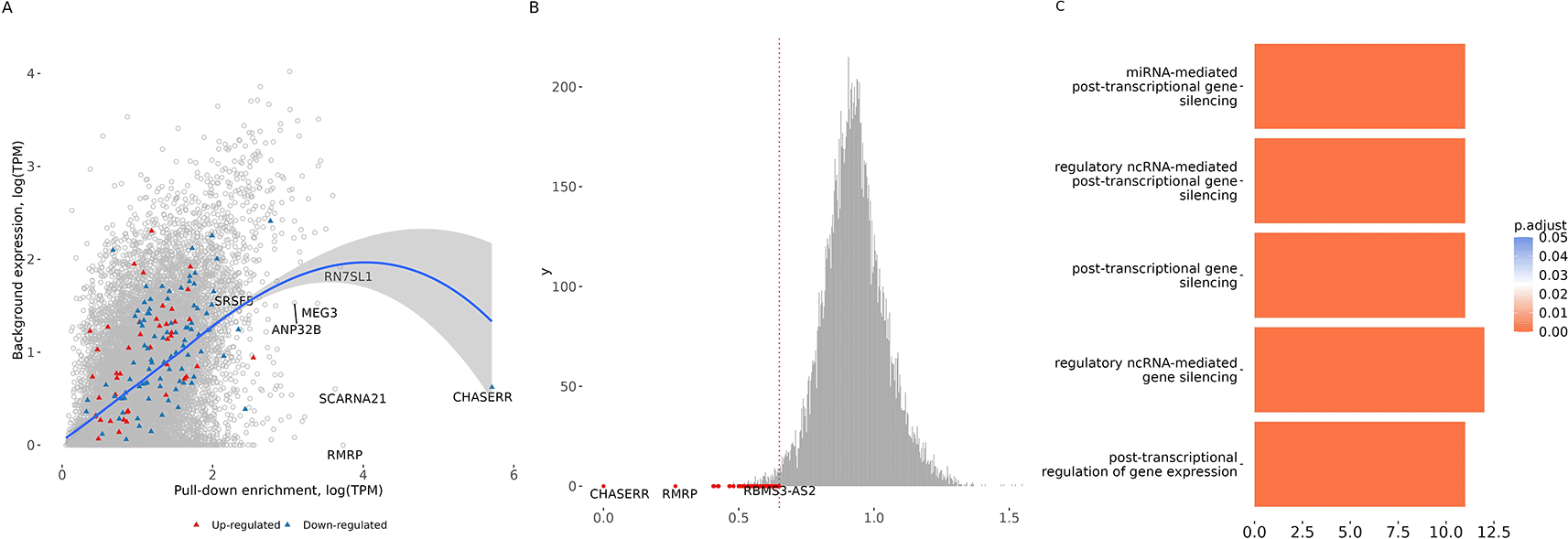
The top-ranking candidate was RMRP, the RNA component of the mitochondrial RNA processing endoribonuclease. The high-confidence interactor set also comprised multiple lncRNAs (including LINC-PINT and FTX) and microRNA host genes (e.g., MIR4435-2HG, MIR100HG), consistent with CHASERR functioning as a regulator for diverse non-coding RNA species. The enrichment in miRNA genes supports prior evidence of CHASERR’s role as a microRNA sponge.10 Gene Ontology enrichment analysis of the network strongly implicated processes of miRNA- and ncRNA-mediated post-transcriptional silencing (Figure 1C), corroborating this molecular role.
To probe functional connectivity, we tested whether interactors were altered upon CHASERR knockdown. The lncRNA CRNDE — the lncRNA that stabilises SIRT1 deacetylase23 — the only interactor, besides CHASERR itself, that was significantly downregulated following knockdown, revealing a specific regulatory connection (FDR < 0.05). Moreover, a direct CHASERR–CRNDE RNA–RNA interaction was predicted computationally using ASSA.13 Together, these findings establish CHASERR as a hub for a defined non-coding RNA network, with RMRP and CRNDE as core components.
We next tested whether CHASERR controls transcription via direct promoter binding. We quantified CHASERR pulldown reads across FANTOM6 promoter regions, normalising to CAGE-seq expression from control fibroblasts. Promoters were stratified based on enrichment: those with a pull-down signal exceeding baseline (normalised expression > 1) and those without (normalised expression < 1).
Promoters enriched for CHASERR binding showed a strong, specific association with transcriptional changes upon its depletion (Figure 2A-E, Supplementary Tables 3, 415). When compared to promoters differentially expressed after CHASERR knockdown, we found a highly significant overlap between upregulated genes and promoters without pull-down signal exceeding baseline (p-value < 2.2e-16 and p-value < 1.056e-05 for two distinct ASOs). Downregulated genes were significantly enriched for CHASERR-bound promoters with pull-down signal exceeding baseline (p-value < 3.371e-14 for ASO 07). Thus, promoter binding by CHASERR is functionally linked to its role in gene regulation.
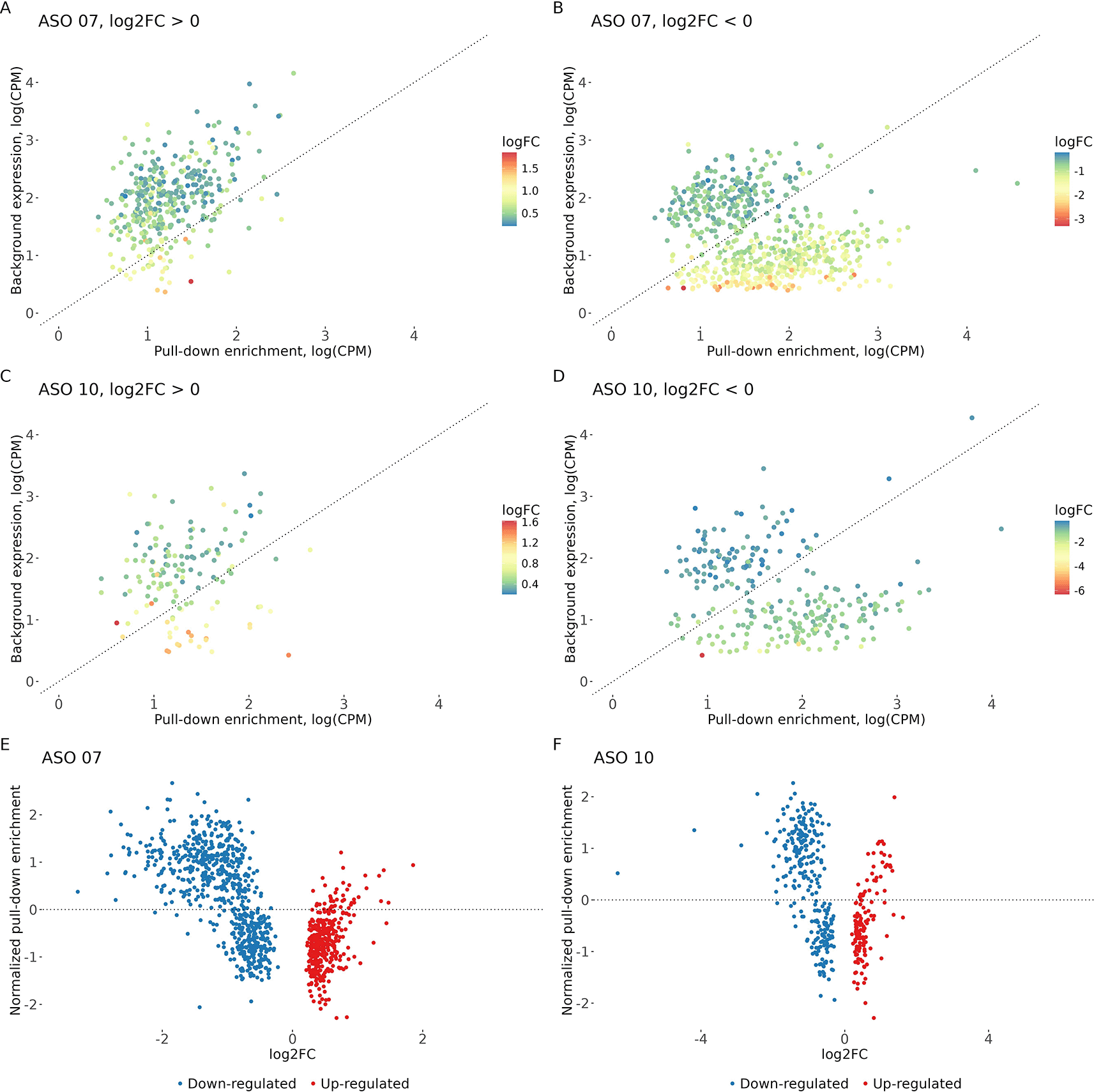
A–D: Correlation between baseline expression and CHASERR pull-down enrichment (log (CPM+1)) for genes that are significantly up- or down-regulated (FDR < 0.05) by two distinct antisense oligonucleotides (ASOs). A, B: ASO 07; C, D: ASO 10. E, F: The magnitude of pull-down enrichment (pull-down – background) correlates with the log2FC for the respective ASOs.
Our findings establish CHASERR as a dual-function regulatory hub, directly tethering to promoters while scaffolding a network of non-coding RNAs. This integrated mechanism explains its broad impact on gene expression and disease. The physical association with microRNA host genes validates its role as a molecular sponge, while promoter binding reveals a direct, previously unknown transcriptional function. The specific, functional link to CRNDE highlights a key regulatory axis within this network.
A central methodological insight is the challenge of distinguishing specific lncRNA interactions from non-specific background. Our co-enrichment-based filter addresses this, but we note that transient or low-affinity interactions may be underrepresented. Furthermore, while our data demonstrate binding and correlation, the precise structural mechanisms of CHASERR’s RNA and DNA engagements, and their spatial coordination in the nucleus, remain to be determined.
Future work must dissect the individual contributions of partners like RMRP and CRNDE and test whether CHASERR’s dual roles are cooperative or context-dependent. Nevertheless, by mapping its physical interactions, we provide a mechanistic blueprint that transforms CHASERR from a disease associated locus into an understood regulatory factor, offering a framework for studying other pleiotropic lncRNAs.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)