Keywords
Vitamin D, binding protein, gene, polymorphisms, Tuberculosis
This article is included in the Genomics and Genetics gateway.
Vitamin D, binding protein, gene, polymorphisms, Tuberculosis
Vitamin D Binding Protein (DBP), also known as group-specific component (Gc), is one of the most prevalent and significant carrier proteins of vitamin D metabolites, accounting for an estimated 85-90% of the total metabolite.1–4 The unbound fraction, which is the free fraction, was estimated to be less than 1%, whereas the albumin-bound fraction was approximately 10-15%.5 DBP, amember of the albumin family, is synthesized in the liver.6 This protein is considered responsible for vitamin D deficiency in target cells, as the bound fraction has a minimal impact on target cells.6,7 Other functions of DBP include actin scavenging, macrophage activation, and fatty acid transport.1
The highly polymorphic DBP gene is located at 4q12-q13, with over 120 variants.8 These genetic variations,affect the circulatory distribution of vitamin D, which leads to vitamin D deficiency.3 In various populations of the world, the two extensively studied non-synonymous DBP single nucleotide polymorphisms (SNPs) rs7041 and rs4588 exhibit variable distributions.2 These variations are located in exon 11, where 7041 encodes c.1296 T>G p.Asp416Glu, while rs4588 encodes c.1307 C>A p.Thr420Lys.9 These two variations give rise to three polymorphic isoforms, which are known to differ by lineage and include Gc1F, Gc1S and Gc2.10,11 The wild type of these SNPs is Gc1Fgenotype variations in the in Gc1F, D416E, and T420K result in, the Gc1S and Gc2 genotypes, respectively.12 Gc2 is found at locus rs4588 while Gc1F and Gc1S are found at locus rs7041.8 Previous studies have shown that people who have the rs7041 G allele as a substitute for the T allele and the rs4588 A allele instead of the C allele have higher levels of DBP and a higher affinity for vitamin D, consequently resulting in lower free and bioavailable vitamin D levels. Consequently, the DBP role controlling total, free, and bioavailable vitamin D is crucial in immunity and influences progression of disease.13
Studies have documented that vitamin D deficiency contributes to TB susceptibility, and individuals with deficiency are at a high risk of developing TB.14 Therefore, vitamin D status is implicated in the response to M. tuberculosis, and is genotype-dependent, varying across geographical areas.15
The wild-type Gc1F genotype is predominantly found in the African population, with a low frequency of Gc2 and Gc1S and is associated with low levels of vitamin D. This association is an effect of DBP concentration levels in different genetic variants. The Gc1F genotype has a low concentration of DBP with high affinity for vitamin D metabolites; consequently, low bioavailable vitamin D levels have been reported.16 Therefore, we performed a cross-sectional study to determine the frequency distribution of DBP gene polymorphisms among ATB patients, LTBI patients, and individuals without TB infection in a Ugandan population.
This was a pilot study based on a previous cross-sectional study of 148 participants between the ages of 12-65 years of which 102 samples were conveniently selected. Details of this previous study have been reported elsewhere.17 This study was nested from a larger study that was conducted in accordance with the Declaration of Helsinki, and approval was granted by the Makerere University School of Biomedical Sciences Higher Degrees Research Ethics Committee (SBS HDREC)/#SBS-637 on 25th Jan 2019, Kiruddu Referral Hospital, and National Council of Science and Technology (HS2639) on the 31st October 2019. All experimental protocols were approved by Makerere University SBS HDREC (#SBS-637) and the National Council of Science and Technology (HS2639), as guided by the Helsinki Declaration. Written informed consent was obtained from active TB patients at Kiruddu Hospital for study participation. Informed consent was obtained from the KTB household contacts, and the parents or guardians consented on behalf of the minors.
Following the inclusion and exclusion criteria, these samples were selected for genotyping of DBP gene polymorphisms. This was based on the availability of whole blood for ATB patients and peripheral blood mononuclear cells (PBMCs) for LTBI patients/individuals and those with no TB infection. After obtaining ethical approval and informed consent, Gen-expert-positive TB patients from Kiruddu Referral Hospital were enrolled, and samples of household contacts of LTBI Individuals with (QFN+ TST+) results and individuals with no TB infectionwho were (QFNTST-) from the Kampala TB (KTB) project were included in the study. Samples from patients with LTBI and those without TB infection were purposively selected. PBMC samples with adequate cellswere selected for genotyping, and samples with fewer cells were excluded. Based on this, 46 samples were excluded becauseof inadequate sample volume and the number of cells available for successful genotyping. Individuals withan HIV+serostatus were not excluded from the study.
The phenol-chloroform (PhCHCL3) method was used to extract DNA from whole blood samples of active TB patients, PBMCs fromLTBIpatients, and those with no TB infection.
Briefly 100 μl of 10% SDS were Dispensed in eppendorf tubes. 150μl of whole blood were then added and mixed by pipeting up and down. This was followed by incubation at 65°C for 10 min using a heat block. 100 μl of 3N Soduim Acetate were added and 5 vortexed vigorously. This was followed by addition of 700 μl of PhCHCL3. And 280 μl of PCR grade water. The tubes were inverted vigorously several times. they were then Centrifuged @ 13000 rpm for 30 min. 450 μl of the aqueous layer was Transferred to a new eppendorf tube. 1000 μl of absolute isopropanol (100%) was then added. DNA was precipitated at -80°C for 20min. this was followed by centrifuging at 14000 rpm for 30 minutes. The isopropanol was removed off leaving approx 50 μl. Add 700 μl of 70% isopropanol were added and Centrifuged @ 14000 rpm for 30 minutes. The 70% isopropanol was completely removed leaving the dry pellet. The DNA tubes were dried at 65°C. DNA was eluted in 100 μl of PCR H2O @ 65°C. It was then stored at – 80 °C for future use.
Agarose gel electrophoresis of human genomic DNA was performed using 1% agarose gel prepared by weighing and dissolving 1.5 g of agarose in 150 ml of 1x TAE (1% solution). The agarose was boiled thoroughly in a microwave oven for 3 minutes to allow thorough heating and mixing, and allowed to cool to 50°C at room temperature. 7.5 μl of 5 mg/ml ethidium bromide was added and mixed well by gentle agitation. The Agarose solution-ethidium bromide mixture was poured into an assembled gel casting tray with a comb attached and allowed to set at room temperature for approximately one hour. Upon setting, the gel was placed in to the electrophoretic tank and the combs vertically removed. 1x TAE buffer was poured in to the electrophoretic tank to just cover the gel. 5 μl of loading dye was added to 5 ul of each of the PCR product on the Para film, mixed and then loaded on to the wells in the gel.
While loading, the molecular weight marker was always loaded on the first lane and then the extracted human genomic DNA. The samples were run at 120 volts (constant voltages, variable current) for 30 mins. After 1 hr, the electrophoresis was stopped and the gel was carefully transferred to a UV trans illuminator for visualization.
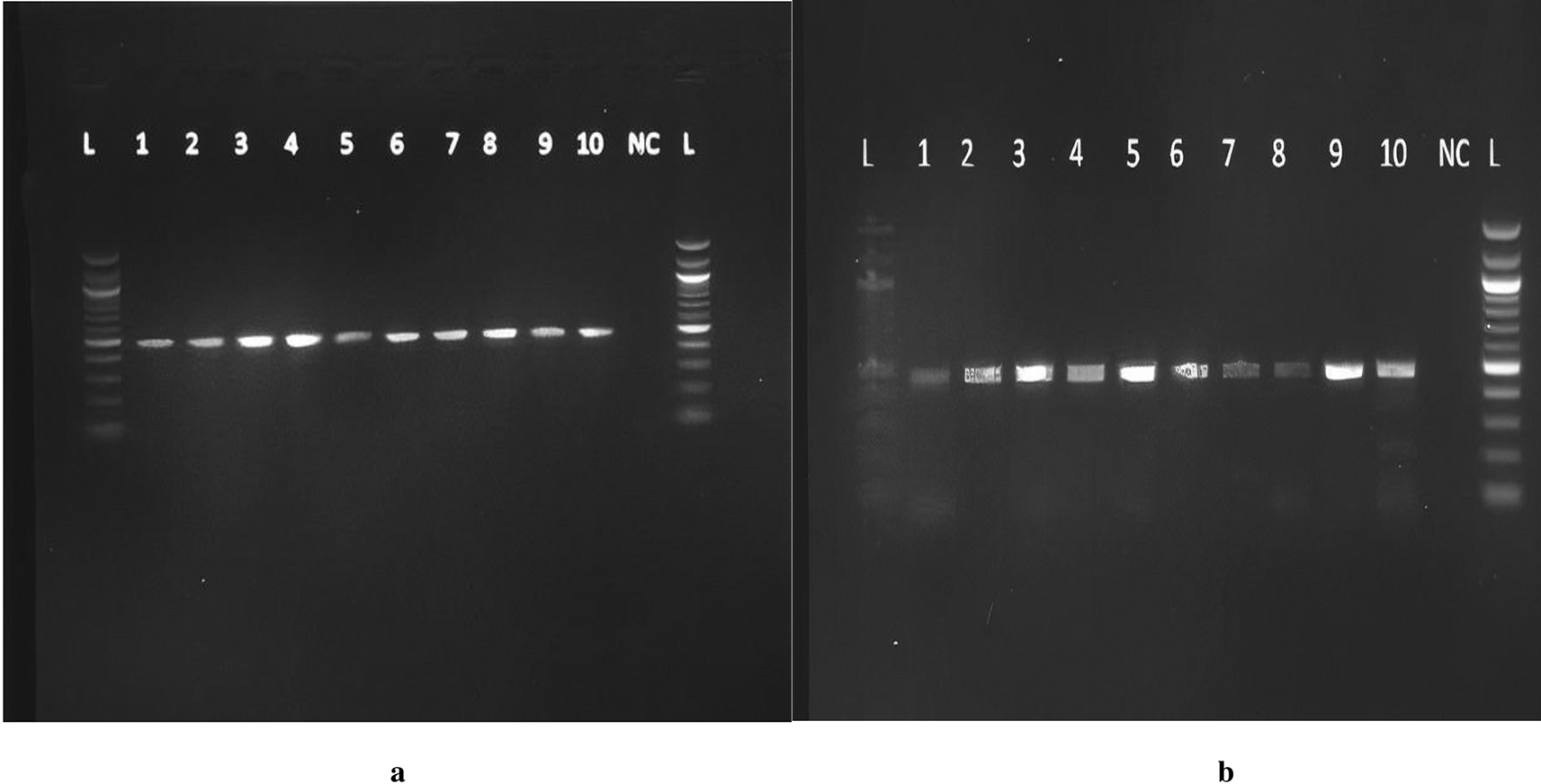
Primers were purchased from Eurofins Genomics, Inc. Germany. The primer sequences were forward 5″AAATAATGAGCAAATGAAAGAAGAC3′ and reverse 5′ CAATAACAGCAAAGAAATGAGTAGA3′ with expected amplicons of approximately 483 bp. Mastermix preparation was performed from the pre-amplification room as follows: 25 μL of 2X Taq Master Mix, 2.5 μl of the reverse primer (6 pM) and 2.5 μl of forward primer (0.6 pM), and 15 μL of PCR water, making a volume of 45 μLforeach reaction. Forty-five microliters of the master mix and 5 μL of DNA were added to each of the PCR tubes. Five microliters of PCR water was added to the negative control tubeand transferred into the SimpliAmp Thermocycler for 40 cycles under the following programmed conditions: enzyme activation step 5 min at 95°C, denaturation for 20 s at 95°C, annealing for 45 s at 56°C, extension for 10 sat 72°C, Final Extension for 5 min at 72°C, and finally an infinite hold at 4°C. The amplicons were run on a 2% agarose gel, as previously described, and a product size of 483 bp was obtained ( Figure 1a and b). Under ambient conditions, the PCR products were sent for Sanger sequencing using the forward primer at ACGT in the United States of America. The ABI Big Dye Termination Kit (Applied Biosystems, USA) and the ABI prism 310 Genetic analyser (Applied Biosystems) was used. The sequenced chromatograms were obtained and cleaned up to remove low yield peaks. A BLAST query sequence was performed to confirm the DBP gene against that of the NCBI library. The gene products were named Homo sapiens Gc vitamin D binding protein (Gc), with sequence sizes between 414 bp and 448 bp with a percent identity of 98-99%. The DBP gene reference sequence (AH004448.2, Homo sapiens vitamin D-binding protein gene) was retrieved from the National Center for Biotechnology Information (NCBI). The raw DBP genesequences from our analysis were aligned to the reference genome. Variant filtering was performed in which low-read regions and errors were identified. Coverage, quality scores, and proximitywere also checked. Sites that differed from the reference genome and sequences were identified and sorted according to their nucleotide and amino acid composition. The detection of the presence of SNPs was performed by searching for the possible change in the codon GAT to GAG at position 416, representing the rs7041 variant, and ACG to AAG at position 420 for the rs4588 variant.
All the figures provided here are only found in a preprint and have not been published anywhere else. I therefore assume I do not need to request for copy right permissions.
The data were summarized using STATA software (Stata Corp. STATA 16.0, College Station, Texas, USA). Frequency and percentage (n [%])were used to determine the frequency distribution of the DBP gene. Pearson’schi-square test was used to compare the frequency of DBP among the study groups and a subanalysis of sex. Fisher’s exact test was used to test for significance among the groups. A potential deviation from Hardy–Weinberg equilibrium was performed using the dnaSP software. V5 (http://www.ub.es/dnasp).
The p-value was considered significant at P < 0.05, with a 95% confidence interval.
Of these, 102 were genotyped, of which 52 were newly diagnosed ATB patients, 23 had LTBI and 27 had no TB infection. The median age of the study participants was 28years, and the majoritywere female (63 [61.2%]). A small proportion of these patients were HIV-positive, 9 (18.4%). Table 1 shows the social, demographic, and clinical characteristics of the study participants and more details of the study participants have been described elsewhere.17
A Gc1S reference sequence with the GAG codon at position 416 was retrieved from the NCBI database (Homo sapiens vitamin D-binding protein gene) for use. According to our search in BioEdit, all our sequences had the wild-type GAT codon at this position compared to the reference sequence. At position 420, all of our samples had an ACG codon, except for two samples that showed a conversion to AAG. Ninety-seven percent of the study population had rs7041 GAT and ACG for the rs4588 codons, 2% had GAT rs7041 and AAG rs4588, and 1% had rs7041 GAG and rs4588 ACG (Gc1S). Figure 2a-c show the details of this analysis and highlighted transformations. Therefore, the frequency distribution of the DBP genotypes in the study population was Gc1F, 97%; Gc, 2.2%; and Gc1S, 1%. The frequency distribution of the DBP genotypes among patients with TB was 96.2% Gc1f, 0% Gc1S, and 3.8% Gc2. Among the LTBI cases, 95.7% were Gc1F, 4.3% were Gc1S, and 0% were Gc2. For those without TB infection, the frequencies were Gc1F 100%, Gc1s 0% and 0% for Gc2. There was no statistically significant difference in the predominant Gc1F genotype among ATB patients, LTBI individuals, and those without TB infection (P=0.3). Notably, the participants with the Gc2 genotype were ATB patients with HIV coinfection. Furthermore, we alsofound that individuals with the Gc1S genotype hadLTBI. The genotype and allele distributions of the study participants are shown in Table 2 and Table 3, respectively. The Hardy-Weinberg equilibrium analysis was in equilibrium, D’=0, P=0.2
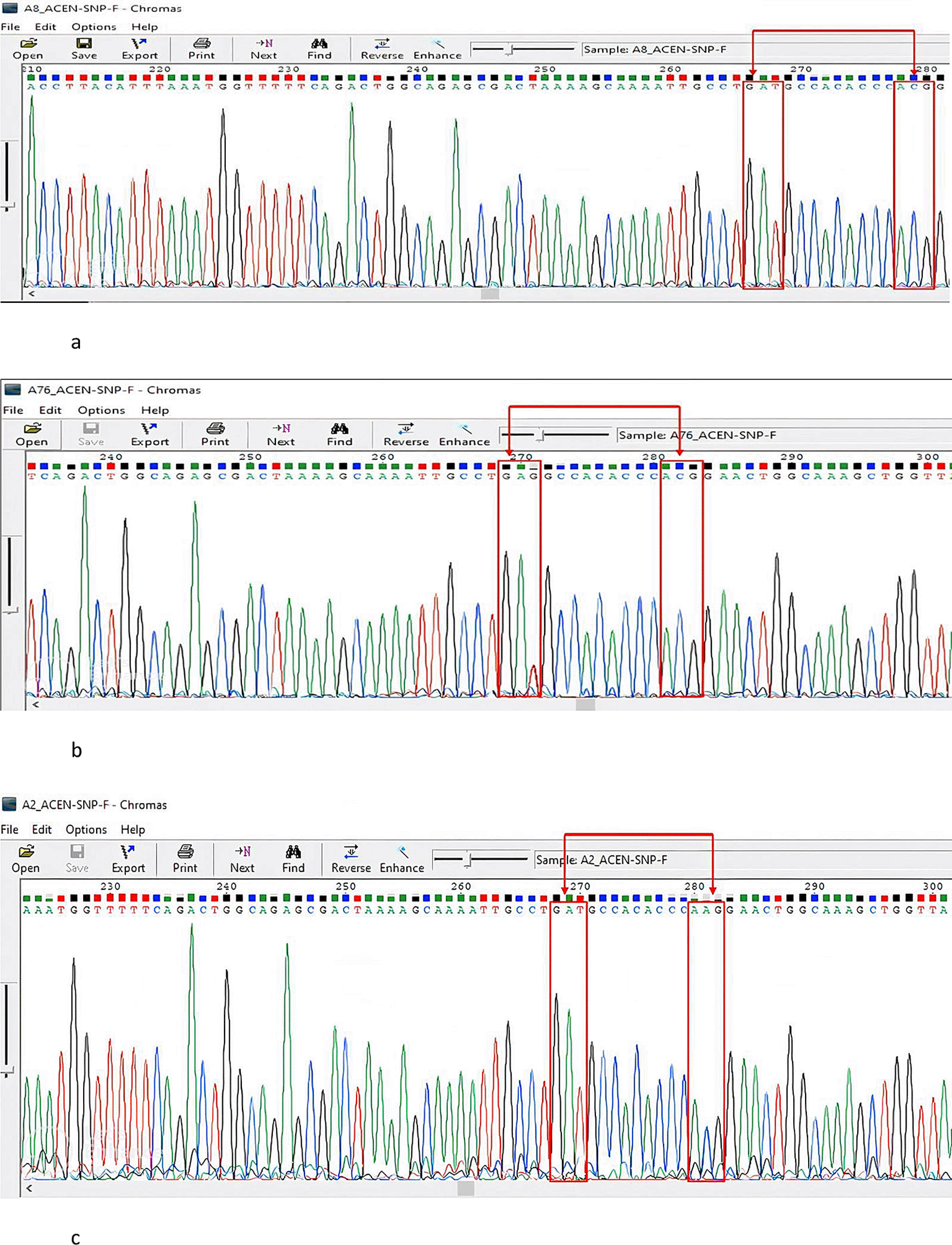
No conversion was observed in both SNPs rs7041and rs4588, The figure was generated using BioEdit 7.2 softwarehttp://www.mbio.ncsu.edu/BioEdit/bioedit.html by A.A. b: A Sanger sequencing representative chromatogram of the GAG and ACG (Gc1S) genotype. A conversion was observed in the rs7041 SNP GAT to GAG and no conversion noted in the rs4588 SNP. The figure was generated using. The figure was generated using BioEdit 7.2, http://www.mbio.ncsu.edu/BioEdit/bioedit.html by A.A. c: A Sanger sequencing representative chromatogram of the GAT and AAG (Gc2)genotype. No conversion was observed in the rs7041 SNP and conversion is noted in the rs4588 SNP from ACG to AAG. The figure was generated using BioEdit 7.2 http://www.mbio.ncsu.edu/BioEdit/bioedit.html by A.A.
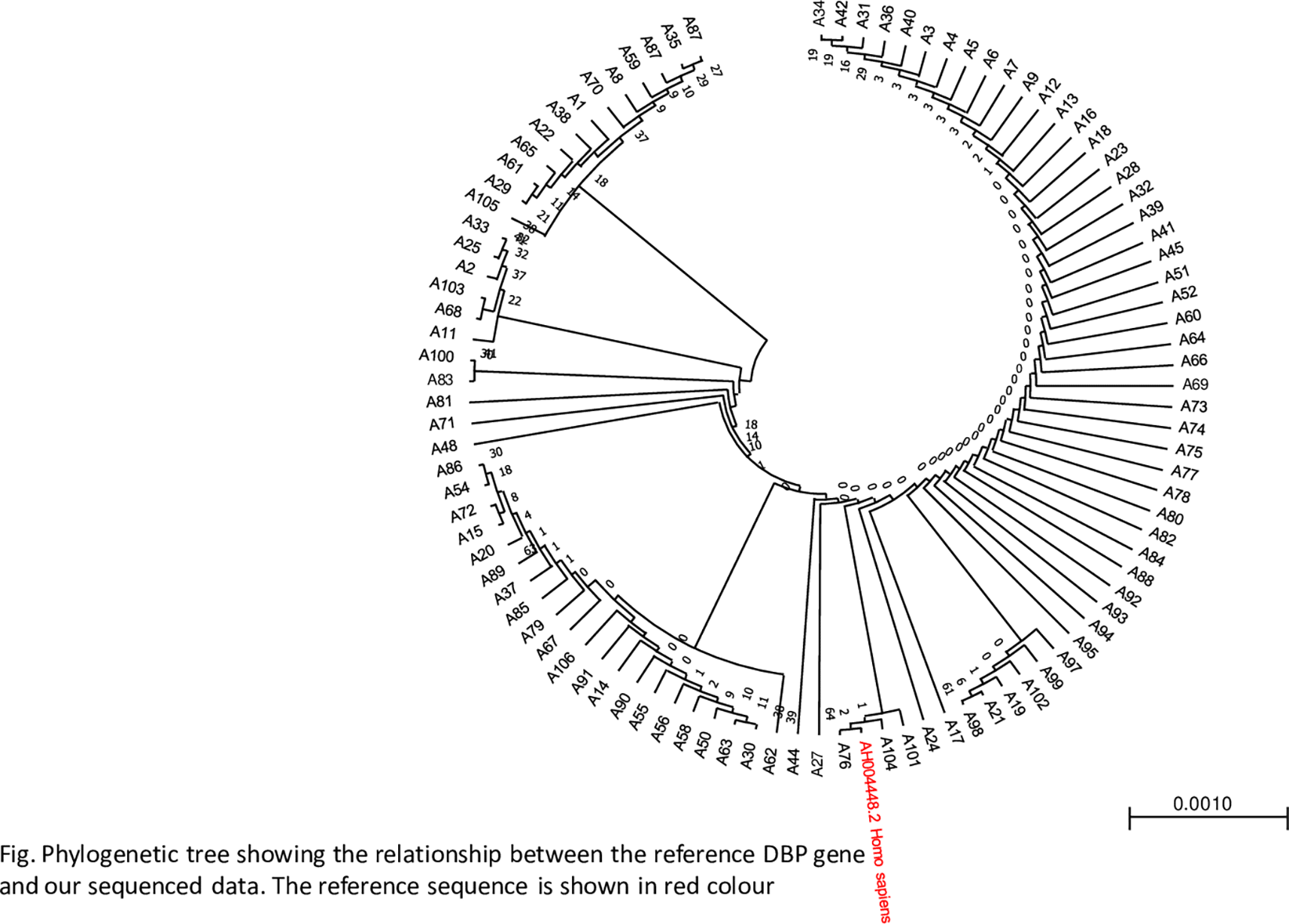
A phylogenetic tree was constructed to determine the closeness of the sequences using the maximum likelihood method. The phylogenetic tree revealed a close relationship between the samples and the reference genes, as shown in Figure 3.
DBP is highly polymorphic, with approximately 120 variants; however, the widely studied variants are the rs7041 and rs4588 SNPs from these three variant genotypes, Gc1F, Gc1S, andGc2. These genotypes arethe predominant source of diversity observed across different geographic locations and ethnicities. Well-documentedreports have focused on multiracial populations and lack adequate information regardinghomogeneous populations. Extensive research in population genetics has found that the frequency of the Gc1F genotype is predominantly found in Africans and African-Americans and that Gc2 is the lowest.18 Our study showed a frequency distribution of 97% of the Gc1F genotype, 2% of the Gc2 genotype, and 1% of the Gc1S genotype in the population. This is consistent with the genotype frequency distribution ofAfrican Black populations. This finding is comparable to that of two West African studies in Gambia, with a nearly homogeneous population like ours. They reported a frequency distribution of 86.0% and 83.3%, respectively, and another study from South Africa reported 80.0%.11,19,20 However, these studies had a larger sample size thanthe current study. Similarly, findings from a study among Black Americans and whites showed a frequency distribution of 92.7% for the Gc1F genotype among Blacks (2.1%) and Gc2 (2%). In contrast, the same study found a high-frequency distribution of Gc1S among the white population.18 In contrast, a study performed among the Eurasian population found the Gc1F genotype to be the lowest (13.7%) and the highest was Gc1S.16 Correspondingly, astudy from Finland reported a low frequency of Gc1F (3.7%).21 The above observations show that DBP polymorphisms are ethnically based; therefore, diverse effects on vitamin D metabolites are likely to be observed. This study did not finda statistically significantdifference in the frequency distribution of the Gc1F DBP genotype among the three study groups (P=0.3). This finding is similar to that of a study from Pakistan that reported a non-significant association of DBP with TB (P=0.3).22 However, we noted that the Gc2 genotype was only found among active TB patients with HIV coinfection. This finding is comparable to that of the previously mentioned South African study that reported an association between the Gc2 genotype and TB status among Asians.23 Furthermore, a recent study in China exploring vitamin D pathwaygene polymorphisms foundthat the DBP Gc2 genotype was associated with progression to pulmonary TB.25 Moreover, in our study, the Gc1S genotype was detectedin the LTBI group. Therefore, these findings suggest that the minor alleles in our population have a genetic association. The Gc1F genotype is predominant in the black population; therefore, it is worth mentioning that our population was consistent with the Hardy-Weinberg equilibrium, and the frequency distribution observed is possibly a representation of our study population. Consequently, in additiontogenetic predisposition, environmental, social, and economic factors may play a major role in TB susceptibility in the population.
Regarding the HIV sub-analysis, no statistical significance was found among the genotypes and TB status.
Considering the analysis of sex and the DBP gene, no statistical difference was observed in the frequency distribution among male and female participants (P=0.07). This observation is similar to thatof a study in India on TB patients.24
We acknowledge that the small sample size and homogeneous population of the current study could have contributed to the less significant effect size needed to detect minor alleles, as observed. Therefore, future studies should considerlarger sample sizes to increase the probability of detecting minor alleles in the population. The strength of this study is that it is the first to determine DBP gene polymorphisms in the Ugandan population of active TB patients, LTBI, and household contacts, providing an adequate representation of TB status.
The frequency distribution of the DBP and Gc1F genotypes was predominantly found in the study population, with no statistically significant difference among the ATB patients, LTBI patients, and those with no TB infection. However, the minor alleles, Gc2 andGc1S, may be associated with a higher risk of active TB and LTBI. Further research is warranted in a larger homogenous and heterogeneous population to adequately determine the functional significance ofminor alleles and their role in TB pathogenesis. This is important for TB control and prevention.
This study was nested from a larger study that was conducted in accordance with the Declaration of Helsinki, and approval was granted bythe Makerere University School of Biomedical Sciences HigherDegrees Research Ethics Committee (SBS HDREC)/#SBS-637 on 25th Jan 2019, Kiruddu Referral Hospital, and National Council of Science and Technology (HS2639) on the 31st October 2019. All experimental protocols were approved by Makerere University SBS HDREC (#SBS-637) and the National Council of Science and Technology (HS2639), as guided by the Helsinki Declaration. Written informed consent was obtained from active TB patients atKiruddu Hospital for study participation. Informed consent was obtained from the KTB household contacts, and the parents or guardians consented on behalf of the minors.
Conceptualization: E.L.A., Data curation: E. L. A., Formal analysis: E. L. A., A. A., R.B. S. N., A. O., Funding Acquisition: E.L.A., D.P. K., Investigation: E.L.A., Project Administration: E.L.A., I.A.B., W.W., D.P.K., M.L.J., Methodology: E. L. A., A. A., R.B.S.N., A.O. K. B., Resources: E.L.A., I, A.B., A.A., R.B. S. N., Software: M. B., A.A. K. B. Supervision: W. W., D.P. K., I. A. B., M L. J., Validation: E.L.A., W. W., D.P. K., I. A. B., M L. J., Visualization: E.L.A., A.A.R.B. S. N., M.B., K.B., A.O., Writing – original draft: E. L. A., Writing – review & editing: All authors reviewed the manuscript.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)