Keywords
Anuran, Hylidae, genome assembly, conservation, Australia, threatened species
This article is included in the Genomics and Genetics gateway.
This article is included in the Galaxy gateway.
Anuran, Hylidae, genome assembly, conservation, Australia, threatened species
Removed duplicate citation and changed Omni-C to Hi-C where possible throughout.
See the authors' detailed response to the review by Yanbo Sun
The alpine tree frog (Litoria verreauxii alpina; Figure 1) is endemic to the Australian Alps of New South Wales and Victoria, occurring at elevations above 1,200 meters (Brannelly et al., 2015). Since the introduction of the pathogenic amphibian chytrid fungus Batrachochytrium dendrobatidis (Bd) to Australia, the species’ distribution has declined by over 80% since the 1980s, leaving only a few remaining populations (Gillespie, Osborne, & McElhinney, 1995; Hunter, Osborne, & Smith, 1998; Osborne, Hunter, & Hollis, 1999; Hunter et al., 2009). Adult L. v. alpina are highly susceptible to Bd infection, with prevalence rates approaching 100% during the breeding season (Brannelly et al., 2015; Scheele et al., 2015). The species exhibits minimal protective immunity against the disease, leading to near-complete population turnover each breeding cycle (Bataille et al., 2015; Grogan et al., 2018; Brannelly et al., 2015; Scheele et al., 2015).

Despite these challenges, the remaining L. v. alpina populations persist, largely due to a compensatory reproductive strategy. Infected individuals exhibit increased reproductive effort, as evidenced by larger gonadal structures and higher gamete production compared to uninfected counterparts (Scheele et al., 2015; Brannelly et al., 2016, 2021, 2025). This strategy may help offset high mortality rates, ensuring continued recruitment despite the overwhelming impact of Bd. However, the long-term effectiveness of this response remains uncertain, particularly as other environmental pressures further threaten population stability. Understanding the genetic mechanisms underlying this reproductive adaptation is crucial for assessing the species’ resilience and informing conservation strategies.
The Litoria genus, to which L. v. alpina belongs, is highly diverse, comprising over 150 recognized species across Australia (‘AmphibiaWeb’, 2025). Despite its ecological and evolutionary significance, no publicly available reference genomes for any species within the genus were identified in the Australian Reference Genome Atlas (ARGA) as of February 25, 2025 (Hall et al., 2023). To address this gap, we generated a high-quality reference genome for L. v. alpina, along with a reproduction-focused transcriptome derived from tissues critical to reproductive function which included the brain, liver, and gonads. These genomic resources provide a foundation for investigating genetic variation within the species, shedding light on how L. v. alpina maintains population persistence in the face of extreme disease pressure. Additionally, this work fills a critical gap in genomic knowledge within Litoria, offering new opportunities to study evolutionary relationships, reproductive adaptations, and broader ecological dynamics across the genus.
Samples were collected from two adult males and one adult female L. v. alpina that were lab-raised from eggs at the University of Melbourne, Werribee campus, Victoria, Australia (Brannelly, Sharma, & Wallace, 2023). The individuals sampled were part of a larger experiment that involved humane euthanasia as the endpoint. Individuals were medically euthanized via immersion for ≥10 min in 3 mL of 100 mg/L tricaine methanesulfonate (MS-222) buffered with sodium bicarbonate. Individuals were removed from the MS-222 solution after becoming unresponsive and immediately decapitated (University of Melbourne’s Animal Ethics application: 26083). Tongue, muscle from the right thigh, and liver tissue were removed from one male (Lva_1) while brain, liver, and gonads (testes or ovaries) were extracted from the other male and female individual (Lva_2 and Lva_3 respectively). All samples were flash frozen using liquid nitrogen and stored at -80°C until extraction.
High molecular weight (HMW) DNA was extracted from the tongue and muscle tissue of Lva_1 using the Monarch® HMW DNA Extraction Kit for Cells & Blood (New England Biolabs: T3050S) following the manufacturer protocols. Concentrations and quality were then assessed via a Femto Pulse genomic DNA 165 kb kit (Agilent: FP-1002-0275), Qubit™ dsDNA BR assay kit (Thermo Fisher Scientific; Table 1), and NanoDrop (Thermo Fisher Scientific; Table 1), with the highest yielding sample used for library preparation.
| Sample | Qubit (ng/μL) | Nanodrop (ng/μL) | 260/280 | 260/230 |
|---|---|---|---|---|
| Lva_1_Muscle | 11.6 | 19.4 | 1.62 | 0.86 |
| Lva_1_Tongue | 376 | 178.1 | 1.79 | 1.88 |
Total RNA was extracted from the brain, liver, and gonad tissues collected from Lva_2 and Lva_3 individuals using the RNAeasy Plus Mini Kit (Qiagen: 74134) with RNAse-free DNAse I set (Qiagen: EN0521) digestion. RNA quantity was determined using a Qubit 3 fluorometer with an Invitrogen™ Qubit RNA High Sensitivity Kit (Thermo Fisher Scientific) and RNA integrity (RIN) score determined using a 5200 Fragment Analyzer (Agilent; Table 2).
The HMW DNA from Lva_1 tongue tissue was sent for Pacific Biosciences High Fidelity (PacBio HiFi) library preparation with a SMRTbell® prep kit 3.0 (Pacific Biosciences: 102-141-700) and Revio™ polymerase kit (Pacific Biosciences: 102-739-100) and sequencing on one single molecule real-time (SMRT) cell on a PacBio Revio at Australian Genome Research Facility (AGRF), Brisbane, Australia.
Two LinkPrep libraries were prepared from liver tissue from Lva_1 using the Dovetail® LinkPrep™ Kit (Cantata Bio) at the Advanced Genomics Services of the Australian Genome Research Facility. Briefly, the chromatin was fixed with disuccinmidyl glutarate (DSG) and formaldehyde in the nucleus. The cross-linked chromatin was then fragmented and tagged with Tn5 transposase in situ. Next, the cells were lysed to extract the chromatin fragments, which were subsequently bound to chromatin capture beads. Proximity ligation was then performed, whereby chromatin fragments that were in proximity to one another were ligated together. After proximity ligation, the crosslinks were reversed, the associated proteins were degraded, and the DNA was purified and converted into a sequencing library. Each library was sequenced on an Illumina Novaseq X plus platform to generate 2 million 2 × 150 bp read pairs to assess the quality of mapping, valid cis - trans reads and complexity of the library. For chromosome level assembly, each library was sequenced approximately 100 million 2 × 150 bp read pairs per Gb of the genome size at the Australian Genome Research Facility, Melbourne, Australia.
Total RNA from the brain, liver, and gonads of individuals Lva_2 and Lva_3 was prepared using Illumina Total RNA with RiboZero Plus library preparation and sequenced as 150 bp paired-end reads on an Illumina NovaSeq 6000 at the Australian Genome Research Facility, Melbourne, Australia.
Genome assembly was conducted on the Galaxy Australia platform using workflows developed by Bioplatforms Australia Threatened Species Initiative, Galaxy Australia, and the Australian BioCommons (https://australianbiocommons.github.io/how-to-guides/). After the upload of the raw HiFi reads in.ccs.bam format provided by AGRF, we used the BAM to FASTQ + QC v1.0 workflow (Price, 2022a). This utilizes SamtoFastq v2.18.2.2 (Broad Institute, 2009), Samtools flagstat v2.0.3 (Danecek et al., 2021), and FastQC v0.72 (Andrews, 2010). Following file conversion, we ran the PacBio HiFi genome assembly using hifiasm v2.1 workflow (Price & Farquharson, 2022). This process produced a draft genome assembly in FASTA format, accompanied by assembly metrics and a detailed report. HiFi reads underwent adapter sequence removal using HiFiAdapterFilt v2.0.0 (Sim et al., 2022), followed by de novo assembly using hifiasm v0.16.1 (Cheng et al., 2021). To evaluate assembly structure and completeness, the assembly graph was visualized using Bandage Image v0.8.1 (Wick et al., 2015). Bandage Info v0.8.1 was used to extract key assembly statistics, such as contig N50 and total assembly length, providing insights into the overall quality of the assembly.
To enhance the assembly’s accuracy, the purge duplicates from hifiasm assembly v1.0 workflow (Price, 2022b) was applied to remove haplotype repeats. This step uses minimap2 v2.28 (Li, 2018) and purge_dups v1.2.6 (Guan et al., 2020) to align and purge duplicates based on read depth. We then scaffolded the Hi-C reads with the genome using the TSI scaffolding with HiC (based on VGP-HiC-scaffolding) v1.0 workflow (Syme & Silver, 2024). The scaffolding workflow utilizes several tools including BWA-MEM2 v2.2.1 (Li & Durbin, 2010; Li, 2013), YAHS v1.21.2 (Zhou, McCarthy, & Durbin, 2023), gfastats v1.3.10 (Formenti et al., 2022), bedtools BAM to BED v2.31.1 (Quinlan & Hall, 2010), and PretextMap v0.1.9 (Harry, n.d.). The finished genome was assessed using the genome assessment post assembly workflow (Farquharson et al., 2024), which produces Fasta statistics v2.0, Quast v5.0.2 (Mikheenko et al., 2018), BUSCO v5.4.6 (Simão et al., 2015), and Merqury v1.3 (Rhie et al., 2020) outputs.
Transcriptome assembly was also conducted on the Galaxy Australia platform using workflows developed by Bioplatforms Australia Threatened Species Initiative. To minimize interference from repetitive genomic elements, the reference genome was first subjected to repeat masking using the Repeat Masking v3.0 workflow (Silver & Syme, 2024a). The workflow processed the reference genome FASTA file, generating both hard-masked and soft-masked genome files along with a statistics report detailing the extent of masking. Quality control and adapter trimming of raw RNA sequencing reads were performed using the QC and Trimming of RNAseq Reads v1.0 workflow (Silver & Syme, 2024b) for each tissue separately. This step involved filtering low-quality bases and removing sequencing adapters. Trimmomatic Galaxy v0.36.6 was used to trim-reads specifying NEXTERA (pair-ended) adapters, SLIDING-WINDOW:4:5, LEADING:5, TRAILING:5 and MINLEN:25 (Bolger, Lohse, & Usadel, 2014). The soft repeat-masked genome was indexed and reads were aligned using HiSAT2 v2.2.1 (Kim et al., 2019). Quality was assessed using FASTQC v0.74 (Andrews, 2010), and the processed reads were retained as paired FASTQ files for subsequent analysis.
Processed RNA-Seq reads were aligned by tissue and individual of origin to the soft-masked reference genome using the Align Reads to Find Transcripts v1.0 workflow (Silver & Syme, 2024c). This alignment generated BAM and GTF files, providing transcript structures and alignment metrics to aid in genome annotation. Transcriptome assembly was conducted using the Combine Transcripts v1.0 workflow (Silver & Syme, 2024d), which integrated tissue-specific transcript data into a comprehensive global transcriptome. Coding sequences were predicted based on sequence homology with Xenopus laevis coding DNA (cDNA) downloaded from NCBI. The workflow output included a GTF file representing the global transcriptome and FASTA sequences of coding transcripts.
To identify the longest isoforms, the Extract Longest Transcripts v1.0 workflow (Silver & Syme, 2024e) was applied. TransDecoder was used to predict coding sequences, filtering transcripts to retain only the longest isoform per gene. The resulting outputs included peptide FASTA files, coding sequence FASTA files, and GFF3 annotation files for further analyses. The final step involved converting the transcriptome annotation outputs into formats compatible with genome annotation tools. The Convert Outputs v1.0 workflow (Silver & Syme, 2024f) was used to process TransDecoder peptide FASTA files and global nucleotide FASTA files into .cdna, .dat, and .pro formats required for downstream annotation applications.
Genome annotation was performed using the FgenesH++ tool on the Galaxy Australia platform using the assembled reference genome, the hard-masked genome and the.cdna, .pro, and.dat files generated by the Convert Outputs v1.0 (Silver & Syme, 2024f) workflows as input files. The Fgenesh annotation v3.0 workflow (Silver, 2024) was executed, which involves genome splitting, annotation, merging of annotation files, and extraction of mRNA, CDS, and protein sequences. The settings used the Xenopus (generic frog) gene-finding matrix and a non-mammalian database. The outputs included GFF3 files of annotated genes and FASTA files for mRNA, CDS, and protein sequences. BUSCO v5.4.6 in ‘protein’ modes was used to assess the annotation with the tetrapoda_odb10 lineage.
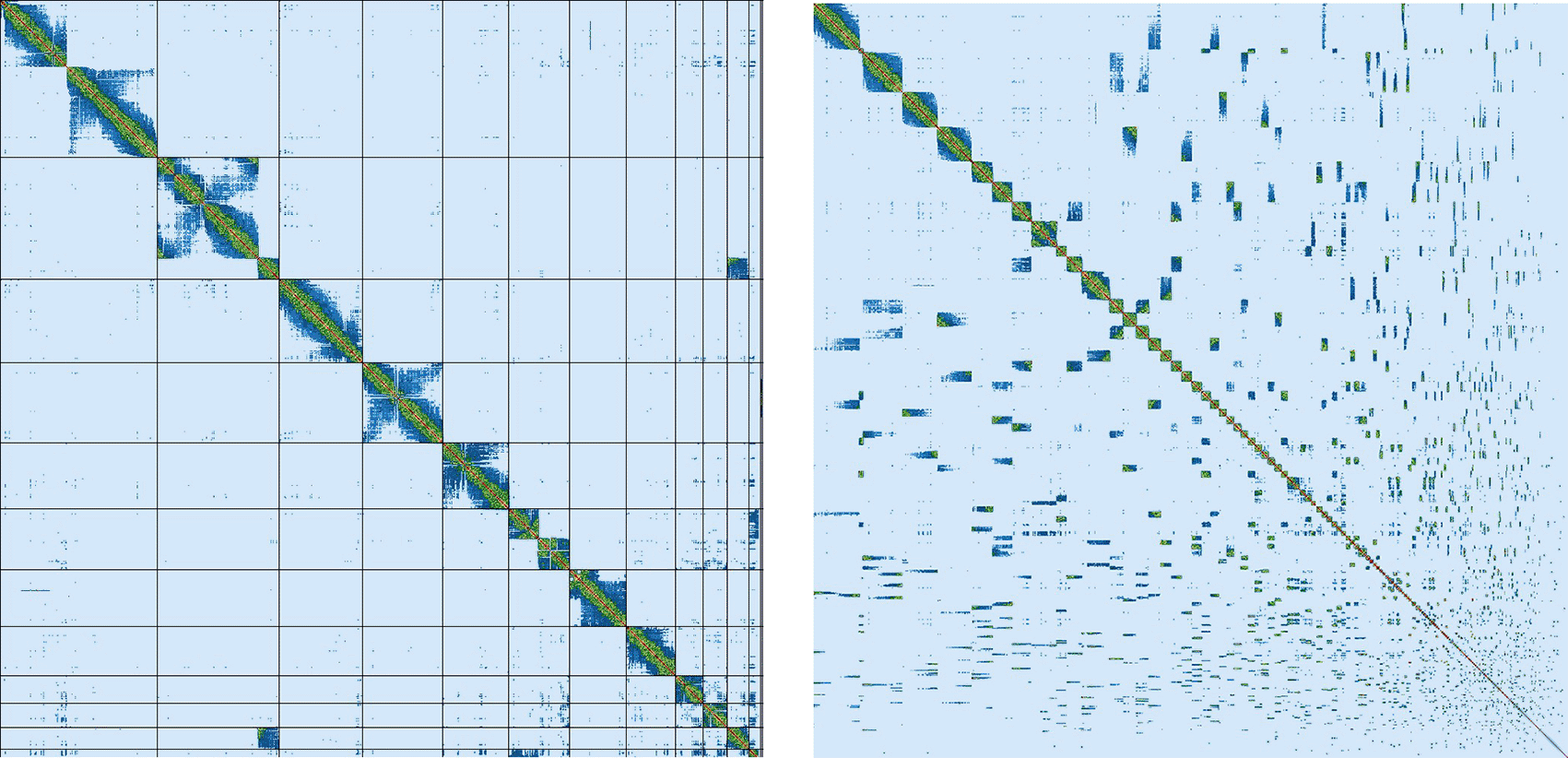
Assembly of the male Litoria verreauxii alpina resulted in a genome of 2.77 Gb, which was comprised of 962 contigs with a contig N50 of 37.17 Mb. The genome was sequenced using PacBio HiFi reads which generated 87.92 Gb from 7,764,356 reads, resulting in a coverage of 31.74×. Primary assembly contigs were scaffolded using proximity-based enrichment Hi-C data ( Figure 2), which produced 248.99 Gb from 829,962,675 reads. Genome scaffolding with this data resulted in 774 scaffolds with 188 gaps and a scaffold N50 of 267.09 Mb ( Table 3). The majority (91.3%) of the assembly mapped to the first 13 scaffolds, reflecting the 13 chromosome karyotype described for the species (Schmid et al., 2018) and within other Litoria species (Ferro et al., 2018; Mollard, Mahony, & West, 2024; Kosch et al., 2025).
The Merqury estimated Quality Value (QV) of the final assembly was 63.7 with an error rate of 4.2e−7. Completeness with Merqury was lower than expected at 78.6%, which is most likely due to the fact that the purge duplicates workflow removed a high number of repetitive sequences and haplotigs ( Figure 3). Before purging, the genome was 2777517237 bp and had a 100% completeness score. After purging, the genome was reduced to 2772442494 bp with most of the purged sequences labeled as high coverage, haplotig, or repeat sequences. BUSCO v5.4.6 indicated a completeness of 90.1% (single = 86.7%, duplicate = 3.4%), using the tetrapoda_odb10 reference set (n = 5310) ( Table 4).
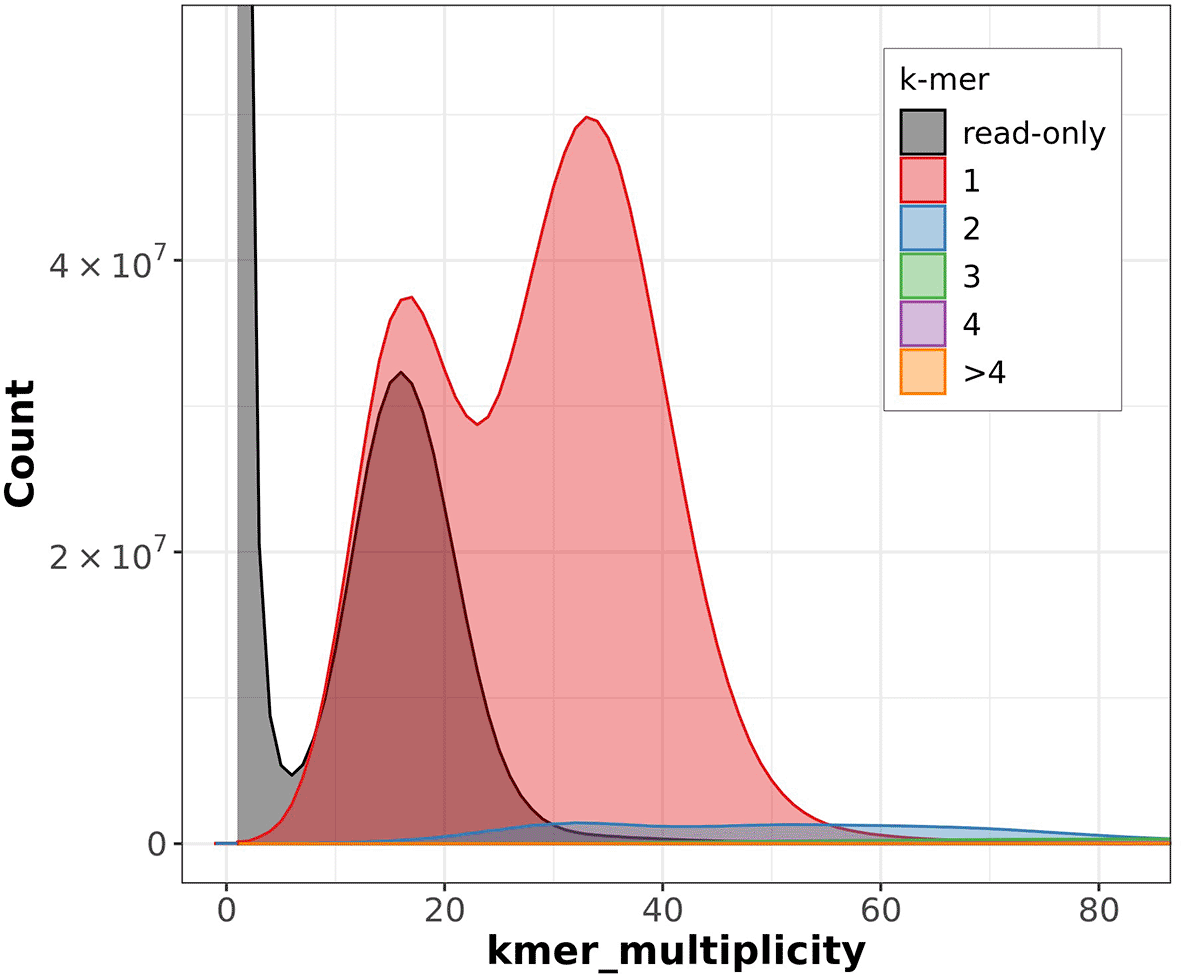
‘k-mer multiplicity’ records the number of times a certain k-mer appears in the reads, and ‘Count’ records the number of k-mers that have appeared that number of times. Grey represents the k-mers found only in the reads, while the colors correspond to the number of k-mers that have appeared at that given number of times.
After quality trimming, 99.48% of reads were retained. Individual tissues had a high number of duplicate reads ranging from 57.2% – 85.0%. The individual tissue transcriptomes had varying mapping rates to the soft repeat-masked genome (78.77% female brain; 77.89% male brain; 80.59% female liver; 83.50% male liver; 80.92% ovary; 81.59% testes). A total of 98760 transcripts were used as evidence for the genome annotation. Repetitive elements comprised 61.43% of the total genomic sequence, with 41.88% of these consisting of unclassified repeats. A total of 40092 genes were predicted from the annotation ( Table 5). There was an average of 6.2 exons (SE=34.6) per putative gene with an average exon length of 229 bp (SE=556) and an average intron length of 3353 bp (SE=12458). The reproduction focused annotation had 65.4% BUSCOs [Single copy: 62.8%; Duplicated: 2.6%]; 14.2% fragmented BUSCOs and 20.4% missing BUSCOs.
| Genome annotation statistics | ||||
|---|---|---|---|---|
| % of genome | Average size (bp) | Median size (bp) | n | |
| Exon | 2 | 229 | 126 | 249568 |
| Gene | 28 | 19299 | 7699 | 40092 |
| Intron | 25 | 3353 | 1033 | 209476 |
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)