Keywords
organoid differentiation, human induced pluripotent stem cells (hiPSCs), 3D pancreatic model, replacement, branching morphogenesis, lineage specification, acinar fate differentiation, pancreatic diseases.
This article is included in the NC3Rs gateway.
organoid differentiation, human induced pluripotent stem cells (hiPSCs), 3D pancreatic model, replacement, branching morphogenesis, lineage specification, acinar fate differentiation, pancreatic diseases.
Scientific benefits
• A protocol for the generation of hiPSC-derived pancreatic organoids results in the induction of branching and formation of distinct acinar and ductal cellular domains.
• A high-throughput approach to interrogate cell signalling.
3Rs benefits
• The protocol promotes the acinar cell lineage without relying on animal-derived matrices for organoid culture.
• Some animal models are used to study gene function in pancreas development, which often requires large numbers of animals to breed and maintain genotypes of interest.
• Better recapitulation of human pancreas development in organoids than in animal models could result in a shift in model usage for lineage-tracing studies.
Practical benefits
• The use of hiPSC lines enables a larger scale of genetic manipulations to be studied and accelerates the timeline for phenotyping compared to mouse experiments.
• The scalability and controllability of organoid models allow more data to be generated via high-throughput experiments, e.g. for genetic/drug screens.
• The burden of genotyping animal models, setting up timed matings, and reducing wastage of animal embryos with incorrect genotypes. Once a specific hiPSC line is generated, it can be frozen and reused.
Current applications
• An in vitro platform for studying pancreatic developmental processes, including cell-cell communication, tissue mechanics, and gene regulation.
Potential applications
The pancreas has two vital functions: endocrine and exocrine, which control digestion and blood glucose levels. The exocrine compartment accounts for over 90% of the organ and acinar cells are the primary constituent.1 They are responsible for producing digestive enzymes, such as amylase, protease, and lipase, which are stored as zymogen granules and released by exocytosis during digestion. Additionally, as part of the exocrine compartment, ductal cells form a ramified network of branches that transport digestive enzymes to the duodenum and release bicarbonate ions to neutralize the acidic contents of the stomach.
During embryogenesis, the pancreas originates from the foregut endoderm at embryonic day (E) 9.5, in mice, and 30 days post-conception (dpc) in humans.2,3 Until E14.5, in mice and 50dpc in human, primary branches form and elongate.4,5 After E14.5, branches start bifurcating and form a ramified network of ducts connecting all the acini to the duodenum. Concomitant with branch formation, pancreatic cells acquire specialized identities and localize to discrete regions of the developing pancreas. Bipotent progenitor cells located in the trunk regions give rise to ductal and endocrine cells, whereas progenitors in the tip regions give rise to acinar cells.6,7 Hence, the cellular mechanisms regulating cell differentiation and branching morphogenesis are spatiotemporally synchronized, so that pancreatic cells organize themselves into a branched architecture fundamental for pancreatic function.
Gene regulatory networks (GRNs) governing pancreatic identities have been extensively characterized in mice and are conserved in humans to some extent.8–10 The transcription factors PTF1A and NKX6.1 respectively govern the allocation of multipotent progenitors to either the acinar fate or ductal/endocrine cell identities.9 However, the roles of many soluble and extracellular matrix proteins in the pancreatic microenvironment remain elusive. The extended time required to study the effects of gene mutations in mice has impeded systematic characterization of developmental signals. Mouse models have been widely employed to study pancreatic development owing to the availability of advanced tools for lineage tracing and gene editing. Although the breeding and maintenance of these models are generally considered mild procedures, they often require large numbers of animals to obtain specific genotypes, collect samples across multiple developmental stages, and ensure experimental reproducibility. Moreover, clear differences have now been described between human and mouse pancreatic development, highlighting the importance of developing human experimental models,11 which would also enable high-throughput functional characterization.
The in vitro differentiation of human induced pluripotent stem cells (hiPSCs) followed by cultures in three dimensions (3D) can generate multicellular structures called organoids, which recapitulate key physiological organ features.12,13 hiPSC-derived organoids enable patient-specific disease modelling and increased genetic diversity compared to non-animal cell sources, such as human embryonic stem cells, for organoid generation. The use of hiPSC-derived organoids enables the systematic targeting of developmental genes using CrispR-Cas9 technology.14 However, most organoid culture protocols rely on animal-derived materials such as Matrigel, which are used as scaffolds to support organoid growth and morphogenesis. Here, we optimized a Matrigel-free 3D culture approach to generate hiPSC-derived pancreatic organoids with branched architecture and proper regionalization of acinar/tip and trunk progenitor compartments.
Understanding what regulates acinar cell differentiation is essential not only for investigating the molecular mechanisms underlying pancreatic development, but also for investigating the underlying pancreatic cell plasticity in disease. Most importantly, acinar cell plasticity is linked to pancreatic inflammation and pancreatic ductal adenocarcinoma (PDAC). During pancreatic inflammation, acinar cells may dedifferentiate into ductal-like progenitor cells that reacquire clear embryonic markers.15 This is a reversible process that allows regeneration of pancreatic tissue after inflammation. However, persistent activation of mutated genes, such as oncogenic Kras, leads to permanent dedifferentiation of acinar cells and is linked to PDAC onset in 98% of cases.1 The animal procedures used to model pancreatic inflammation and PDAC range from moderate to severe.16,17 Besides replacing the use of animal procedures, human pancreatic organoids hold great promise for drug screening purposes. The organoid models developed previously either lacked a physiologically relevant architecture or still required transplantation in mice, and thus remained low-throughput and dependent on animal surgical procedures.18 Hence, we anticipate that the physiological relevance of the pancreatic organoid platform developed in this study could support the development of high-throughput preclinical studies, while replacing mouse models of pancreatic diseases.
hiPSCs (AICS-0090-39)19 were obtained from the Coriell Institute for Medical Research (CIRM) and maintained according to the manufacturer’s protocol (https://www.coriell.org/0/PDF/Allen/ipsc/AICS-0090-391_CofA.pdf) in Matrigel-coated (Corning) 6 well-plates with mTesr1 growth medium (STEMCELL Technologies) supplemented with 1% penicillin-streptomycin (P/S) in a humidified incubator (37°C, 5% CO2). hiPSCs were thawed at 37°C in a water bath, and the cell suspension was then transferred dropwise to a Falcon tube containing enough growth medium for a 1:10 dilution. hiPSCs were centrifuged at 300 × RCF for 5 min, resuspended in growth medium containing 5μM Rho-associated protein kinase (ROCK) inhibitor, and then seeded onto Matrigel-coated plates for maintenance.
Cells were passaged as single-cells at 70-80% confluency using Accutase (Invitrogen) and medium supplemented with 5μM ROCK inhibitor Y-27632 (Sigma) on the day of passaging. Passages of hiPSCs exhibiting significant background differentiation were excluded from the differentiation experiments, and hiPSCs were passaged at least once after thawing and prior to commencing differentiation. The number of passages used for all experiments was less than 45. Cryopreservation of hiPSCs was carried out in freezing medium comprising 60% mTesr1, 30% knockout serum replacement (KoSR), and 10% dimethyl sulphoxide for 24h at -80°C using controlled-rate freezing. The cells were then transferred to liquid nitrogen for long-term storage.
hiPSCs were seeded at a density of 200,000-300,000 cells/cm2 in Matrigel-coated 6-well plates and grown for 24h in mTesr1 supplemented with 1% P/S and 5 μM ROCK inhibitor. Cells were subsequently differentiated in a medium that was replenished daily and supplemented with cytokines to induce their differentiation into pancreatic progenitors. The cytokine and small compound concentrations used were as described by Barsby et al.20
Briefly, the first Basal Medium (basal medium 1) used between days (D) 1 and 5 of differentiation was composed of MCDB131 medium supplemented with Glutamax (1x), glucose (2.5 M), NaHCO3 (1.5g/l), and BSA (0.5%). The second Basal Medium (basal medium 2) used between D6 and the end of differentiation was made of MCDB131 medium supplemented with Glutamax (1x), glucose (2.5 M), NaHCO3 (2.5g/l), BSA (2%), and ITS-X (0.5x). The reagents used are listed in Table 1.
The supplements used to differentiate the pancreatic progenitors were iteratively added to the basal media, as detailed in Table 2.
On D11 of differentiation, pancreatic progenitors (PP) were dissociated using TrypLE (Gibco) and left to aggregate in the Aggrewell microwell system (STEMCELL Technologies) for 72h. Progenitors were seeded in Aggrewell plates at a density of 1200 cells/microwell. After PP clusters had formed in the Aggrewell (72h), they were transferred to suspension plates, and the basal medium was supplemented with the cytokines and small compounds listed in Table 3.
Here, we describe a step-by-step protocol for the generation and culture of pancreatic organoids derived from pancreatic progenitors cultured in 6-well plates and collected on D11 of differentiation. The general steps of cell dissociation, aggregation, and suspension culture were similar to those described by Barsby et al. However, the timing and culture media have been modified and optimized to induce exocrine differentiation and branching morphogenesis, instead of endocrine differentiation. The reagents used in these steps are listed in Table 2.
Preparation of the Aggrewell microwell system (24-well plate)
- Add 500 μL of anti-adherence rinsing solution to each well and centrifuge at 1300 RCF for 5 min.
- The solution was removed by aspirating the anti-adherence solution and add 500 μL of MCDB131 medium.
Dissociation and collection of pancreatic progenitors
- Wash pancreatic progenitors (cultured in a 6-well plate) by adding 1 ml of versene to each well and removing it after 1 min.
- Add 1ml of TrypLE to each well and incubate for 8min at 37°C.
- Keep at 37°C until mild-tapping of the plate causes cell detachment.
Add 1 ml of MCDB131 medium per well and gently flush until most cells are detached.
Add another 1 ml of MCDB131 medium per well and collect cells from all wells into a 50 ml tube.
- Count cells
Centrifuge the 50 ml tube with the cell suspension at 300 RCF for 3 min.
- Aspirate the supernatant and gently resuspend the cells in pre-warmed Basal Medium 2 supplemented with cytokines and small compounds listed in Table 3 (D11-D13).
- MCDB131 medium was aspirated from the Aggrewell plate, replace with 2mL D11-D13 medium, and pancreatic progenitors were seeded at a density of 1200 cells/microwell.
- Centrifuge the plate at 100 g for 3 min.
- Incubate plates in a humidified cell culture incubator (37°C, 5%CO2).
Media changes
- Carefully remove 1 ml of medium from each well by keeping the plate horizontal and aspirating slowly with a pipette tip while taking care not to dislodge clusters at the base of the well.
Add 1 ml of fresh pre-warmed D11-D13 medium gently on the top side of the wells.
Prepare D14 culture medium
On this day, cell clusters collected from 2 wells of the Aggrewell plate are pooled in one ultra-low attachment (ULA) well of a 6-well plate.
- Prepare enough D14-D15 medium (Basal Medium 2 with supplements listed in Table 3) to add 5 ml per well of the ULA 6-well plate.
Transfer of pancreatic progenitor clusters to suspension culture
- For each well of the Aggrewell plate, 1 ml of medium and gently flushed out from the edge of the well to dislodge cell clusters from the microwells.
Coat the pipette tip used to transfer the clusters to the medium.
Swirl the Aggrewell plate to pool the cell clusters toward the center of the well and transfer them into a ULA plate. Pool cell clusters were collected from two wells of the Aggrewell plate in one ULA well.
Once all cell clusters have been transferred into the ultra-low attachment plate, swirl this plate gently in one direction to pool the clusters into the center.
- Remove most of the medium by ensuring that cell clusters are not aspirated, which should be visually visible.
- Add 5ml of D14-D15 medium per well.
- Place the plate in a humidified cell incubator (37°C, 5% CO2) on a rotating platform (100RPM).
- Swirl the ULA plate to center cell clusters in the wells.
- Check the formation of branches as a marker for efficient organoid maturation.
- Remove 4 ml of medium by aspirating from the well edge and replacing it with fresh D16-D18 medium.
- Place the plate back into a humidified cell incubator (37°C, 5% CO2) on a rotating platform (100RPM).
Experiments were performed using pancreatic organoids or following the steps below to fix the organoids.
Immunostaining of cells
In addition to the 6-well plates used to generate pancreatic progenitor-derived organoids, hiPSCs were also cultured in 24-well plates in 1 ml differentiation medium at a cell seeding density of 200,000-300,000 cells/cm2. These plates were used to confirm the efficiency of hiPSC differentiation into pancreatic progenitors by immunostaining for acinar and ductal progenitor markers at D14. Cells were washed in 1ml PBS, fixed for 20 min at 4°C with 4 % paraformaldehyde, and washed again. Fixed cells were then blocked for 45min in a blocking buffer composed of PBS, Donkey serum (Sigma) (3%), and Triton-X (0.1%). Primary antibodies were added to the blocking buffer at dilutions listed in Table 4 and incubated overnight at 4°C. The next day, cells were washed multiple times in PBS with Triton-X (0.1%) and incubated for 1 h in the dark with the secondary antibodies diluted in blocking buffer ( Table 4) and counterstained with Hoechst. Before imaging, the cells were washed multiple times with PBS.
Immunostaining of organoids
Throughout the staining procedure, the solutions were changed by centrifuging the organoids at 200 g for 2 min, removing the supernatant, and adding the next solution.
First, fixed organoids were blocked for 24h in a blocking buffer made from PBS, Donkey serum (3%), and Triton-X (0.5%) at 4°C on a rotating platform. Primary antibodies were added to the blocking buffer at the dilutions listed in Table 4 and incubated for 24h under similar conditions. The next day, organoids were washed three times for 45min in PBS with Triton-X (0.1%), and then incubated for 24h in the dark at 4°C on a rotating platform with the secondary antibodies diluted in blocking buffer ( Table 4), and counterstained with Hoechst. Before imaging, organoids were washed twice in PBS with Triton-X (0.1%) for 45min and once in PBS.
Imaging and image analysis
Immunostained organoids were placed in a 96-well plate with a glass bottom and imaged using a Zeiss LSM 700 confocal microscope. Using ImageJ,21 we quantified cell numbers using the in-built cell counter tool and measured PDX1 staining intensity levels in the nuclei by creating a mask on the Hoechst channel. PDX1 intensity was quantified by measuring the mean pixel intensity in the region of interest (ROI) delineating the AMY+ and NKX6.1+ cells.
RT-qPCR
Cells from the planar culture were dissociated and harvested with TrypLE, and organoids/cell clusters were collected with a trimmed pipette tip pre-coated with 1mL of medium previously removed from the well. These were centrifuged for 3 min at 300 RCF or for 2 min at 200 RCF, respectively. The supernatant was aspirated, and the pellets were washed once with PBS (and then frozen at -80°C). A High Pure RNA Isolation Kit (Roche) was used for RNA extraction and all samples were treated with DNase to remove genomic DNA. cDNA was synthesized using a Transcriptor First Strand cDNA Synthesis Kit (Roche). RT-qPCR was performed on a LightCycler (Roche) with FastStart Essential SYBR Green Master Mix (Roche). The 2-ΔΔCt method22 was used to evaluate the relative gene expression, and the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for normalization. The cDNA dilution was modified to return Cts of approximately 20 for GAPDH, and missing Ct values were set to 40 when the threshold for detectable fluorescence was not met. The primers used for RT-qPCR are listed in Table 5.
Quantification and statistical analysis
All statistical analyses were performed using the R package rstatix. The elements shown in the boxplots are the 25% (Q1, upper box boundary), 50% (median, black line within the boxes), and 75% (Q3, lower box boundary) quartiles, and the whiskers represent a maximum of 1.5X × interquartile range. P-values were calculated using a two-tailed t-test. Normality was assessed using the Shapiro-Wilk normality test. Statistical significance was calculated using unpaired Student’s t-tests for data with normal distributions, and Mann-Whitney U-tests otherwise.
hiPSCs were differentiated into pancreatic progenitors as described by Barsby et al.20 with minor modifications, which are outlined in the Materials and Methods (Figure 1A). The efficiency of pancreatic progenitor differentiation was monitored by assessing the morphology of differentiating cells with a phase contrast light microscope between day (D8) and D10 of differentiation and by immunostaining for the pancreatic progenitor markers PDX1 and NKX6.1, at D11 of differentiation (Figure 1B). Pancreatic progenitors were predominantly double-positive for PDX1 and NKX6.1, and clusters were efficiently formed within 24 h in the Aggrewell plate.
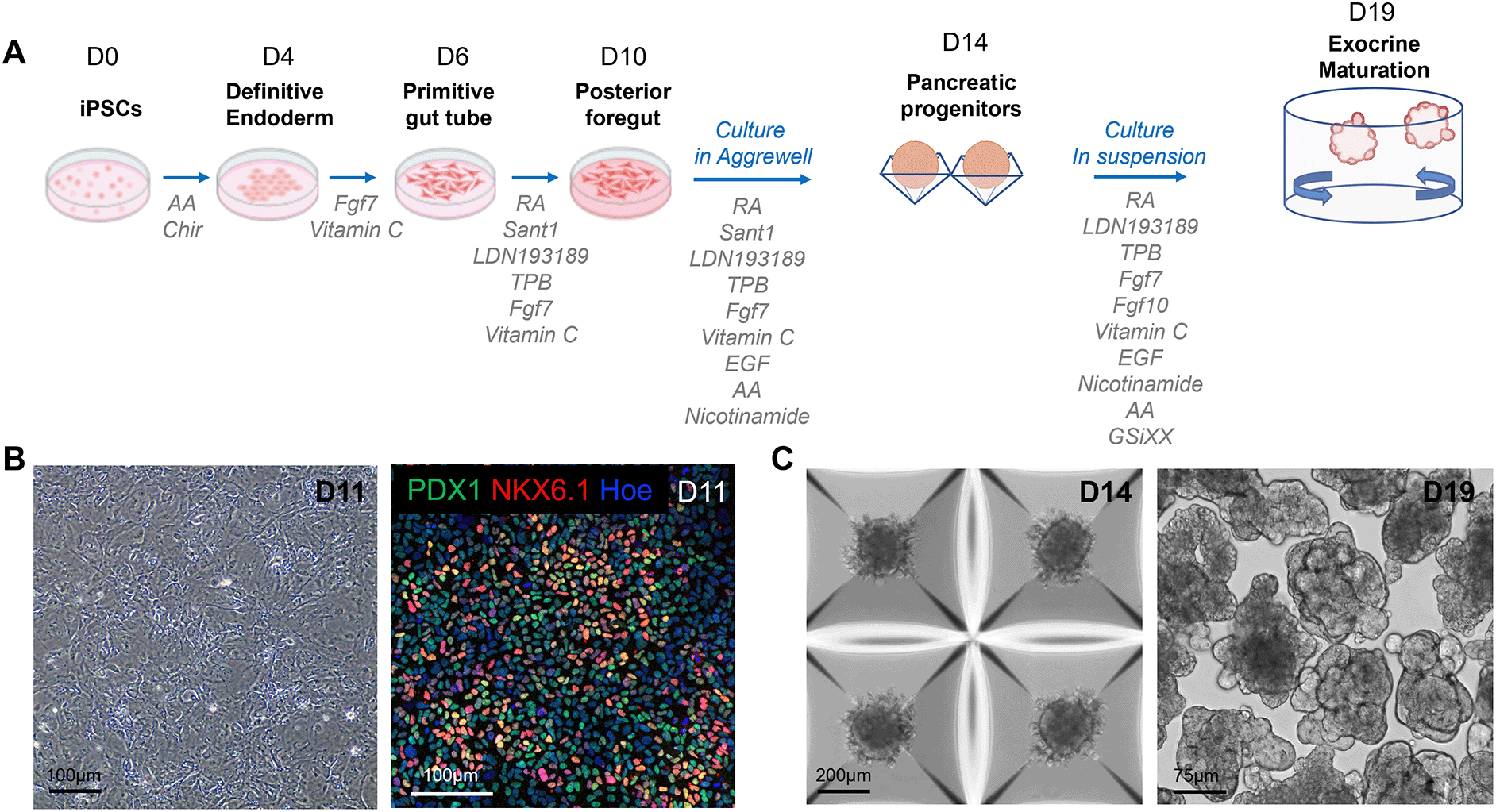
A. Schematic overview and media composition for the differentiation of hiPSCs into pancreatic organoids. The differentiation was carried out as a monolayer until day 11 (D11), then transferred to the Aggrewell microwell system from D12 to D14, and finally performed in suspension until D19. B. Representative phase contrast image (left panel) and confocal image (right panel) of D11 pancreatic progenitors immunostained with the PDX1 and NKX6.1 pancreatic markers. C. Representative phase contrast images of pancreatic clusters cultured in the Aggrewell microwell system on D14 of the differentiation (left panel) and in suspension on D19 of the differentiation (right panel). AA: Activin A, RA: Retinoic Acid.
The dissociated pancreatic progenitors were left to aggregate in Aggrewell plates (Figure 1C). After progenitor clusters were formed, they were transferred to suspension plates for the final stage of differentiation, using an optimized differentiation medium (see composition in Table 3). Specifically, we supplemented the medium with GSiXX, a Notch inhibitor known to promote pancreatic cell differentiation,23,24 along with FGF7 and FGF10, which stimulate the growth and morphogenesis of the exocrine pancreas.25 In contrast to Barsby et al.,20 we excluded factors known to induce endocrine differentiation, such as thyroid hormone receptor agonists, TGF-β antagonists, and betacellulin. This medium, combined with the suspension culture format, triggered primary branch formation in organoids between D17 and D18. 93.5% (SD = 2.3) of organoids acquired a branched architecture by D19 of the differentiation, highlighting a high efficiency of morphogenesis (Figure 1C; 2A).
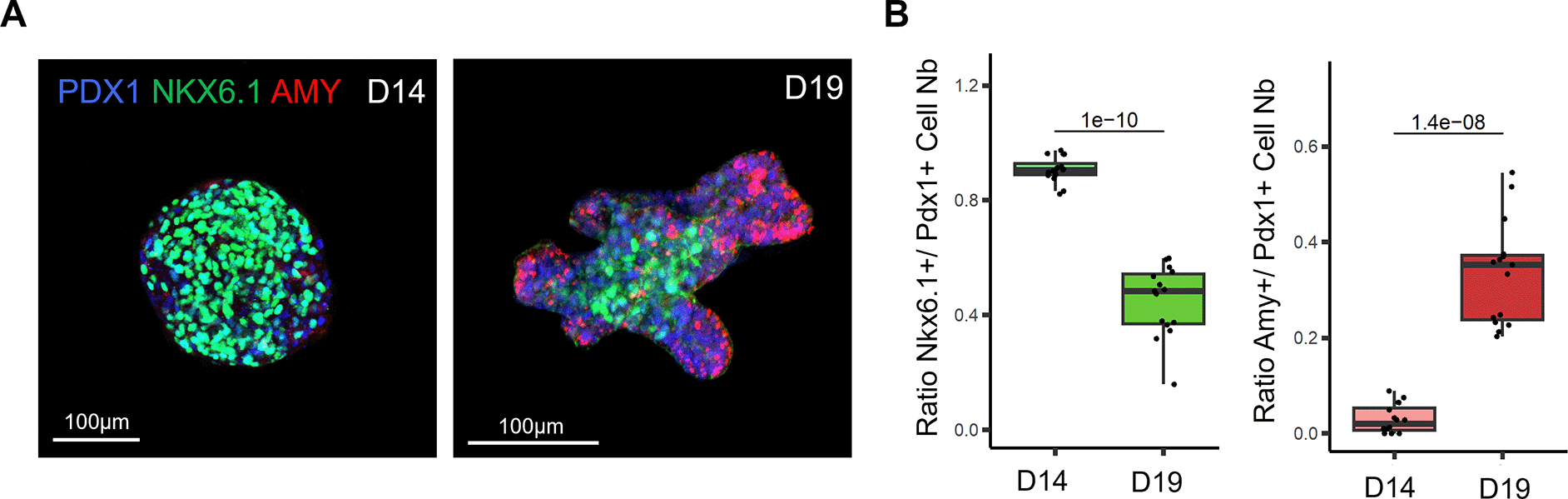
A. Representative confocal images of D14 pancreatic clusters (left panel) and D19 pancreatic organoids (right panel) immunostained for PDX1, NKX6.1 and Amylase (AMY), which respectively mark all pancreatic progenitor cells, trunk progenitor cells and acinar cells. B. Ratio of PDX1+ cells positive for the trunk marker NKX6.1 at D14 and D19 (left panel). Ratio of PDX1+ cells positive for acinar marker Amy at D14 and D19 (right panel). N=13-15 organoids per stage analysed, from N=3 independent differentiation experiments. P-values generated from two-tailed t-tests are displayed on the boxplots.
To investigate the efficiency of our culture medium to induce exocrine differentiation in organoids, we performed immunostaining for tip and trunk markers (Figure 2A). Our whole-mount immunostaining highlights that the stalk region of the organoid branches is positively marked by the NKX6.1 trunk marker, whereas the tip regions are marked by the amylase acinar marker. Hence, the pattern of expression of both these markers is mutually exclusive and clearly defines the tip and trunk domains in organoids. In addition, our quantification showed that the acinar domain strongly increased in the last stage of differentiation between D14 and D19, demonstrating that our protocol supports acinar cell differentiation (Figure 2B). Altogether, our differentiation protocol recapitulated the patterning of the pancreatic branches observed in vivo, with acinar cells positioned at the tip of the branches.
It has been previously shown that during in vivo pancreatic development, PDX1 expression levels decrease in differentiating acinar cells.26 To confirm that this process was recapitulated in our organoid system, we quantified PDX1 staining intensity in AMY+ acinar cells compared to NKX6.1+ cells in organoids collected on D19 (Figure 3A). Our results showed that the PDX1 staining intensity was significantly lower in AMY+ acinar cells than in NKX6.1+ cells at this stage, thus recapitulating the decrease in PDX1 expression observed in vivo.
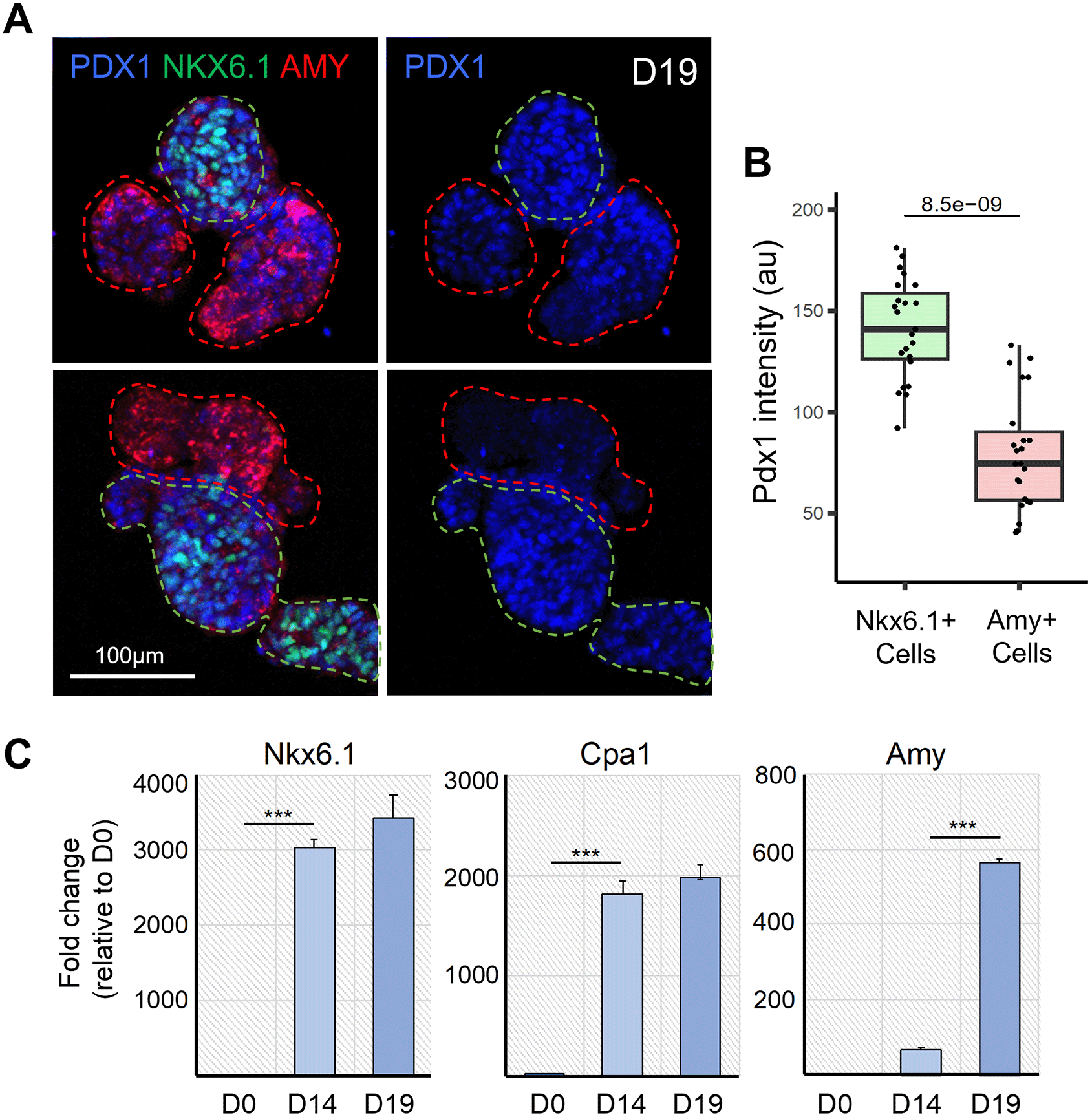
A. Representative confocal images of D19 pancreatic organoids immunostained for PDX1, NKX6.1 and AMY, which respectively mark all pancreatic progenitor cells, trunk bipotent cells and acinar cells (left panel). PDX1 is shown alone in the right panel. Dotted green and red lines respectively delineate the NKX6.1+ trunk and AMY+ tip regions. B. Quantification of PDX1 staining intensity in the NKX6.1+ and AMY+ cells. N = 17-22 organoids analysed, from N = 3 independent differentiation experiments. The p-value generated from two-tailed t-test is displayed on the boxplot. C. Representative qRT-PCR analysis (N = 1 shown) for the indicated pancreatic genes performed on cells, clusters and organoids collected at D0, D14 and D19 of the differentiation protocol. Data are represented as relative fold change. Values shown are mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; two-tailed paired t tests.
In addition, we performed RT-qPCR on dissociated cells collected at D0, D14, and D19 to investigate the efficiency of our protocol in inducing cell differentiation, specifically the acinar fate. Our results highlight that differentiating cells acquire a pancreatic fate by D14, as evidenced by the strong expression of the pancreatic progenitor markers NKX6.1 and CPA1 (Figure 3B). Between D14 and D19, we observed that the level of Amylase expression was strongly increased in organoids, and CPA1 expression was also maintained, indicating that our organoid culture medium promoted the differentiation of acinar cells. Altogether, the differentiation of acinar cells and the formation of a branched architecture demonstrated that our protocol enables the formation of pancreatic organoids with an exocrine identity.
The derivation of pancreatic organoids from either stem cells or primary tissue allows the possibility of replicating native pancreatic organization, morphology, and the emergence of distinct cell populations in health and disease.27 The cell sources of human pancreatic organoids include hPSCs, primary cells from adult tissues, and fetal tissue obtained between 7-11 wpc.13 Human PSC-derived pancreatic organoids are capable of giving rise to ductal or acinar cells that usually self-organize as polarized spheres.28,29 However, the generation of human stem cell-derived pancreatic organoids with a proper branched architecture and correct patterning of the acinar/tip and trunk cell domains has been an ongoing challenge.
The advent of pancreatic organoids for use as an in vitro model has followed many years of optimization of pancreatic cell differentiation protocols,30–32 which aim to mimic pancreatic development. Most recently, a highly optimized protocol was developed20 that incorporated several existing protocols and specifically used SHH and BMP inhibition, FGF and RA signalling activation, and the discovery that joint administration of Nicotinamide and Epidermal Growth Factor (EGF) can increase pancreatic progenitor cell yield.33 Nicotinamide has been shown to increase the proportion of PDX1+NKX6.1+ cells by inhibiting CK1 and ROCK,34 while EGF is known to activate the MAP kinase pathway and is required for pancreatic progenitor proliferation.35 This combination resulted in a significantly improved yield of PDX1+NKX6.1+ pancreatic progenitors compared to established protocols.20,36 Incorporation of this protocol was essential for our work, as it has also been reported that a differentiation efficiency resulting in fewer than 60% PDX1+NKX6.1+ pancreatic progenitors typically does not form aggregates of sufficient size when using the Aggrewell platform. This result also highlights the importance of adequately replicating the in vivo complexity to achieve effective production of pancreatic cell types with high fidelity.
Here, we present a protocol for the generation of hiPSC-derived pancreatic organoids that resemble the neonatal pancreas. We have shown that these organoids harbor regionalized tip and trunk domains. Prominent branching and organoid expansion were observed during the final stage of differentiation, which may have resulted from supplementation with Fgf7 and Fgf10, which are important for the growth and morphogenesis of the exocrine pancreas.37,38 Fgf10 is also known to interact with and activate Notch to maintain pancreatic progenitor cell self-renewal.38–40 However, Notch suppression is required for proper lineage diversification and specification,23,41 and several protocols use media supplemented with Notch inhibitors for the differentiation of PSCs into different pancreatic cell types in vitro.31,42,43 Since in this study, Notch inhibitor was applied from D14 onwards, our findings suggest that regionalization is at least in part achieved by D14, after which Notch inhibition enables regionalized cell differentiation. The use of the Aggrewell microwell platform was a necessary intermediary step in creating an environment that facilitated cell interactions in 3D, without which branched organoid structures could not be generated. Moreover, our protocol eliminates the need for matrices such as Matrigel or collagen to culture pancreatic organoids. This is a significant advantage, as it reduces the use of animal-derived reagents and minimizes the unpredictability of organoid differentiation efficiency due to reagent batch-to-batch variability.
Further optimization will be needed to determine whether the maturation of these organoids can be improved when cultured for extended periods of time and if they can undergo directed differentiation into endocrine cell types. An in vitro model that replicates pancreatic morphology and harbors all pancreatic cell types would equip scientists to answer questions on how branching morphogenesis is coupled with cell differentiation and the role of neighboring and rare cell types during human pancreas development. Endocrine cell differentiation in these organoids would also facilitate research on monogenic forms of diabetes and type 1 and 2 diabetes (T1/2D) using patient-derived cell lines. In the case of T1D, a co-culture system consisting of patient-derived pancreatic organoids and immune cells could also provide a means to study patient-specific beta-immune cell interactions.
The fact that pancreatic cells dedifferentiate into embryonic-like states during pancreatic inflammation and PDAC,15 which are the states replicated in our system, suggests that our organoid platform could allow in vitro modelling of many aspects of these diseases and enable preclinical investigations in a high-throughput setup while replacing animal models. Pancreatic organoids derived from pluripotent human stem cells have been used to study PDAC initiation and progression in culture by induction of KRAS and GNAS oncogenes.29,44 To date, these studies have been performed using cystic organoids composed solely of ductal or acinar cells. Inducing the expression of KRAS and GNAS oncogenes in our organoid platform would enable the research community to study the effects of these mutations in a system that recapitulates the branched architecture of the pancreas and the patterning of its acinar and ductal compartments. Moreover, the high-throughput nature of this platform could allow the screening of drugs that reduce tissue alterations caused by oncogene induction, and thus identify potential therapeutic targets for pancreatitis and PDAC.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)