Keywords
transcriptome, quantification, assembly, discovery, annotation
This article is included in the Cell & Molecular Biology gateway.
This article is included in the Nanopore Analysis gateway.
transcriptome, quantification, assembly, discovery, annotation
Direct RNA and cDNA sequencing technologies from Oxford Nanopore Technologies (ONT) produce long transcriptome reads with high yields at relatively low cost. However, the per-base error rates of ONT reads are still much higher than those of Illumina reads. Several computational tools have recently been developed to assemble transcripts and quantify isoforms in samples sequenced using ONT reads, including FLAIR (Tang AD et al., 2020), ESPRESSO (Gao Y et al., 2023), and IsoQuant (Prjibelski AD et al., 2023). All these tools begin by mapping the long reads to the genome using the Minimap2 (Li H, 2018) aligner in spliced alignment mode. However, the high error rate of ONT reads makes it challenging to precisely identify splice sites through spliced alignment alone. Therefore, these tools incorporate additional information to locate the splice sites accurately. FLAIR can correctly identify splice sites by either using alignments of short-read RNA-seq data or by using a reference annotation. ESPRESSO accepts novel splice junctions only if at least one read aligns perfectly to the reference genome within 10 nucleotides (nt) of the splice site, a stringent criterion that limits its ability to discover novel junctions. IsoQuant replaces novel splice sites with nearby annotated sites within a user-defined distance and restores short, skipped exons according to the reference annotation. For all these programs, misalignments can lead to incorrect identification of splice junctions, which may subsequently result in inaccurate transcript reconstruction.
Here, we present NIFFLR (Novel IsoForm Finder using Long Reads), a tool designed to construct and quantify both annotated and novel isoforms using a reference annotation and long RNA sequencing reads. Unlike other isoform identification tools, NIFFLR does not rely on a spliced aligner to map reads onto the reference genome. Instead, it extracts exons from the given annotation and aligns them directly to the long reads. NIFFLR then constructs transcripts by identifying an optimal path through the mapped exons for each long read, removes redundant transcripts that are contained within others, filters out transcripts with low read support, compares the predicted transcripts to the reference annotation, and finally quantifies both annotated and novel isoforms. For efficient exon-to-read alignment, NIFFLR uses a custom aligner based on a partial suffix array adapted from the MaSuRCA assembler (Zimin et al., 2013).
We designed the NIFFLR algorithm to build transcripts (i.e., sequences of exons) by computing the optimal tiling of every long read using exons and transcripts provided as input. We require the following inputs: long RNA sequencing reads in FASTQ format, a reference genome sequence file in FASTA format, and a reference annotation file in GTF format.
First, we extract the exon sequences from the reference genome using the annotation and output them into a FASTA file. The name of each exon encodes the chromosome name, start and end position on the chromosome, the name of the gene to which the exon belongs, and its orientation. We reverse complement all exon sequences that are on the reverse strand.
We then use a version of a technique first utilized in the MaSuRCA assembler (Zimin et al., 2013) to efficiently compute approximate alignments of exons to the long reads. This alignment technique, which we refer to as psa_aligner, is based on a partial suffix array (PSA). The PSA is designed to efficiently compute approximate alignments, or alignment intervals between two sets of DNA sequences. The psa_aligner first builds a partial suffix array from a concatenated string S containing the sequences of all exons, separated by the letter ‘N’ (note that no ‘N’ characters are allowed in the reference sequence). We also record the starting position of each exon in S. Unlike a traditional suffix array, the PSA limits the suffix size to a predefined value K. The suffix array allows us to quickly locate all occurrences of a given subsequence of length K (or a K-mer) within S, and thus identify all exons and positions where a particular K-mer occurs. We then examine each K-mer in a given long read and compute all the longest common sub-sequences (LCS) of K-mers between the read and the exons, using a default value of K = 12. The approximate alignment coordinates are then determined by calculating the best linear fit between the positions of K-mers belonging to the LCS in the read and on the exon. We only retain alignments where matching K-mers cover at least 35% of the bases within the match interval. Each alignment provides alignment start and end positions, along with the exon and read overhangs, as shown in Figure 1. For each exon, we record the number of K-mers in the LCS, the alignment start and end positions, and the implied start and end on the read. The implied start is calculated as alignment_start-a_overhang, and the implied alignment end is alignment_stop+b_overhang.
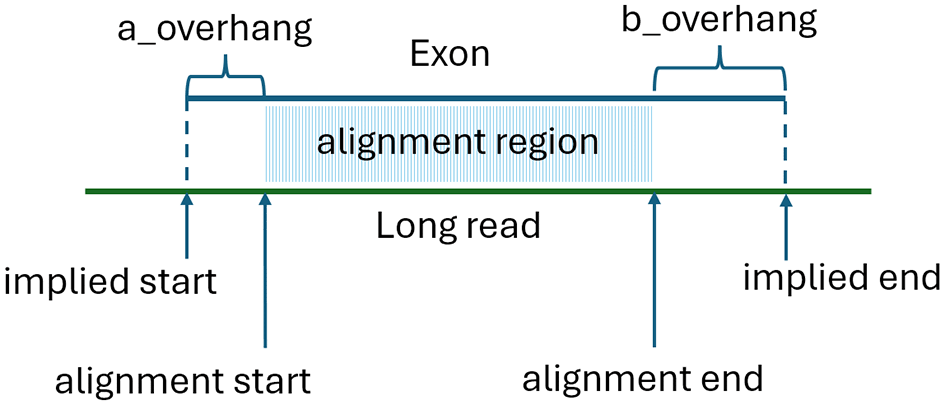
After building the alignments, we assign each long read to a gene locus using a “majority vote” approach. Specifically, for each read, we compute the total number of K-mers in all LCSs for all matching exons from different gene loci and assign the read to the locus L whose exons have the highest total number of matching K-mers. Alignments of any exons that belong to different gene loci are then discarded. Next, we build the transcript matching the read by finding the best tiling of read using exons that belong to locus L. The best sequence maximizes coverage of the read while minimizing gaps or overlaps in the implied alignment coordinates. The long read defines a 5’ to 3’ forward direction, specifying a topological order. We sort the aligning exons in the order of their “alignment start” coordinates if aligned in the forward direction, or “alignment end” coordinates if aligned in the reverse direction. Since we only kept alignments of exons that all belong to a single gene locus L, the exons must all align either in forward or reverse direction. For simplicity, below we describe the algorithm assuming all exons are aligned in the forward direction; the reverse case is treated the same, by reversing the long read.
We represent the exon tiling problem as a graph, where nodes represent exons and edges are defined by gaps or overlaps of 20 bases or less between the implied end of an exon and the implied start of the following exon in the topological order. Next, we choose the “starting” nodes that are not connected on the left. A starting node must be connected on the right and have an “alignment start” closest to the 5’ end of the long read or fully cover it. If multiple exons share the same “alignment start” coordinate due to alternative splicing, we select the exon with the smallest 5’ “overhang”. If the 5’ overhang is the same for more than one exon, we use all such exons as alternative start nodes. We solve the exon tiling problem by finding the longest path through the graph, starting from any start node that minimizes the penalty, defined as the average gap/overlap size between connected exons in the path. In case of a tie, we select the path that maximizes the sum of exon matching lengths minus the sum of the overhangs of the first and last exons. Figure 2 illustrates an example of such a path. Once the longest path is identified, we examine the genomic coordinates of the exons, which are encoded in their sequence IDs. We eliminate the path if there is an overlap between the genomic coordinates of the exons in the path, which could indicate that the long read is chimeric or that there is a significant local genome rearrangement that NIFFLR cannot handle.
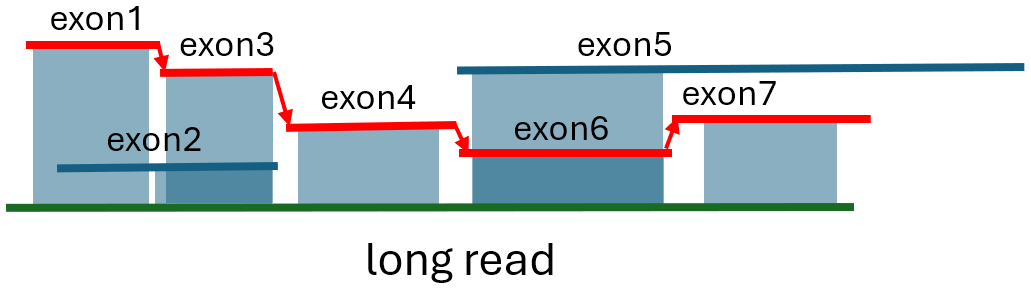
Shading shows the alignment regions. Arrows indicate links. The best path shown in red is the longest path that minimizes the gap/overlap/overhang penalty. Exon1 is chosen as the start exon because exon1+ exon3 have a longer alignment than exon2. Exon5 is alternatively spliced compared to exon6 and exon7, and its longest match is the same as exon6’s, shorter than exon6 and exon7 combined, and hence not selected for the optimal path. Exon2 is alternatively spliced as well.
We convert the best path of exons for each read into a plausible transcript and then group reads that yield the same transcript. For each transcript, we record the reads contributing to it, along with the minimum of the average gap/overlap penalty (Amin) and the minimum of the maximum gap/overlap penalty (Gmin) across all paths of reads that yielded the transcript. In subsequent steps, we use only those transcripts where Amin < 5 and Gmin < 15. These values are empirically obtained parameters and they yielded the best performance in our experiments with simulated reads.
We then use the GffCompare tool to create a set of maximal transcripts by removing those whose intron chains are contained in longer assembled transcripts. We call this set of transcripts “non-redundant”. Next, we perform the first round of transcript quantification, using all originally assembled transcripts to assign reads to the non-redundant transcripts based on containment. Reads from assembled transcripts, which are contained in multiple maximal transcripts, are distributed proportionally to the size of the container transcripts. For each maximal transcript, we calculate the following:
1. The number of reads supporting the transcript.
2. The minimum read coverage across all intron junctions.
3. The total number of junctions covered by at least one read.
4. The portion of the transcript covered by reads.
By design, all intron junctions are covered in the maximal set. After quantification, we perform a transcript recovery step where we attempt to recover reference annotation transcripts that are likely present in the sample, but their intron chains are not completely covered by any long read. If a maximal transcript is contained within a reference transcript, we tentatively replace the contained transcript with the containing transcript from the reference. We then perform quantification again and eliminate multi-exon reference transcripts where none of the intron junctions are spanned by long reads, which means that only one exon had reads aligned to it. These reference transcripts are unlikely to be present in the sample. This procedure is designed to eliminate computed isoforms whose intron chains are contained in the reference transcripts, as these are unlikely to represent genuinely novel isoforms and are likely sequenced from known transcripts. Next, we identify novel transcripts (i.e., those not present in the reference) and apply stricter filtering criteria, requiring the minimum average gap in the exon paths to be less than 2 and the minimum of the maximum gap to be less than 5. This yields the final set of transcripts, containing both novel and known transcripts, which we then again quantify to produce the final set of quantified transcripts.
NIFFLR is designed to run under 64-bit Linux operating system. NIFFLR requires at least 16Gb of RAM and supports multi-core multi-threaded hardware environment. NIFFLR code consists of shell and Python scipts and C++ code. We provide installation instructions for NIFFLR on github: https://github.com/alguoo314/NIFFLR. Basic usage of NIFFLR is as follows: /path/nifflr.sh -r genome.fasta -f reads.fastq -g genome.gtf.
In this section, we compare NIFFLR to other similar methods such as FLAIR2, IsoQuant, and ESPRESSO, and discuss the results of applying NIFFLR to ONT data from the Genotype-Tissue Expression (GTEx) project (Glinos et al., 2022). We performed two evaluations to compare NIFFLR to the existing methods. First, we assessed the performance of each program on a set of simulated ONT direct RNA sequencing reads. Next, we tested all programs on a sample from the GTEx project that was sequenced using both Illumina and ONT technologies.
We simulated reads using NanoSim software (Yang et al., 2017) from the human reference genome GRCh38.p14 and its corresponding RefSeq genome annotation (RS_2024_08). We derived read error profiles from ONT reads of GTEx sample 1192X, which was sequenced with both Illumina RNA-seq and ONT technologies. We used the Illumina reads from the same sample to generate an expression profile for the simulation. Our simulated data set contained approximately 7.8 million reads with an average error rate of 8.7% and an N50 read length of 944 bp. According to Nanosim output, the simulated set had 50,748 unique transcripts expressed.
All programs in this comparison allow the use of a reference annotation to identify and correct splice junctions, and we provided such annotation in all our experiments. Note that FLAIR and IsoQuant have options allowing them to run without annotation, but their accuracy is higher if annotation is provided. To make the evaluation more realistic, we split the reference annotation into a “core” set of transcripts, which is the set with the smallest number of transcripts where each exon was present at least once (referred to as the known set), and the rest of the transcripts (referred to as the novel set). By design, the core set contained every reference donor and acceptor splice site at least once. We provided the core set but not the novel set to all programs. This way we ensured that some portion of the expressed transcripts were not present in the input set of the reference transcripts, enabling us to measure the programs’ ability to discover and quantify novel transcripts in addition to the known transcripts. Our simulated set consisted of reads simulated from 50,748 transcripts, of which 33,686 comprised the core set and the remaining 17,062 comprised the novel set. In our experiments, we measured the number of novel and known transcripts correctly recovered by the programs, as well as the number of false positive transcripts, using the GffCompare tool (Pertea & Pertea, 2020). False positives were defined as any transcripts output by the programs that did not have a complete intron chain match to a transcript in the known or novel set. Table 1 shows the comparison of the programs on the simulated data. NIFFLR has the best sensitivity in recovering known, novel, and all isoforms, and the best overall F1 score, while only losing to IsoQuant in precision. NIFFLR recovers the most isoforms from both the known and novel sets while keeping the number of spurious isoforms relatively low. This result demonstrates that when novel isoform discovery and quantification are the primary goals, NIFFLR is the best tool.
The best values are in bold. NIFFLR recovers the most novel isoforms and the most isoforms total (32,711) while keeping the number of erroneous isoforms lower than FLAIR2 and ESPRESSO, resulting in the best sensitivity and F1 score for isoform recovery. Isoquant is the most conservative and the least sensitive, both on novel and known isoform discovery.
We compared the read counts computed by each program for every transcript to the actual counts from the simulation. Figure 3a presents box-and-whisker plots of the ratios (expressed as base-2-logarithms) of the actual and computed counts for each transcript. The box spans the upper and lower quartile of the ratios and the whiskers represent the range for 95% of the values, with individual outliers outside of the 95% interval shown as dots. NIFFLR has a tighter distribution than FLAIR and ESPRESSO, though it is slightly outperformed by IsoQuant. ESPRESSO shows the worst overall performance, both in terms of the distribution’s tightness and bias. Figure 3b shows a more detailed comparison of the ratios between the computed counts from NIFFLR and IsoQuant, compared to the actual counts for the subset of 18,686 isoforms quantified by both tools. We observe that in this comparison the accuracy is nearly identical, with NIFFLR counts showing less overall bias. This figure suggests that the reason for the slightly lower accuracy (wider whiskers) of NIFFLR compared to IsoQuant in panel (a) is the inclusion of counts for many more transcripts by NIFFLR, capturing less reliable lower-count transcripts, which IsoQuant discards. In the simulated data comparison, NIFFLR demonstrates superior quantification accuracy and sensitivity overall.
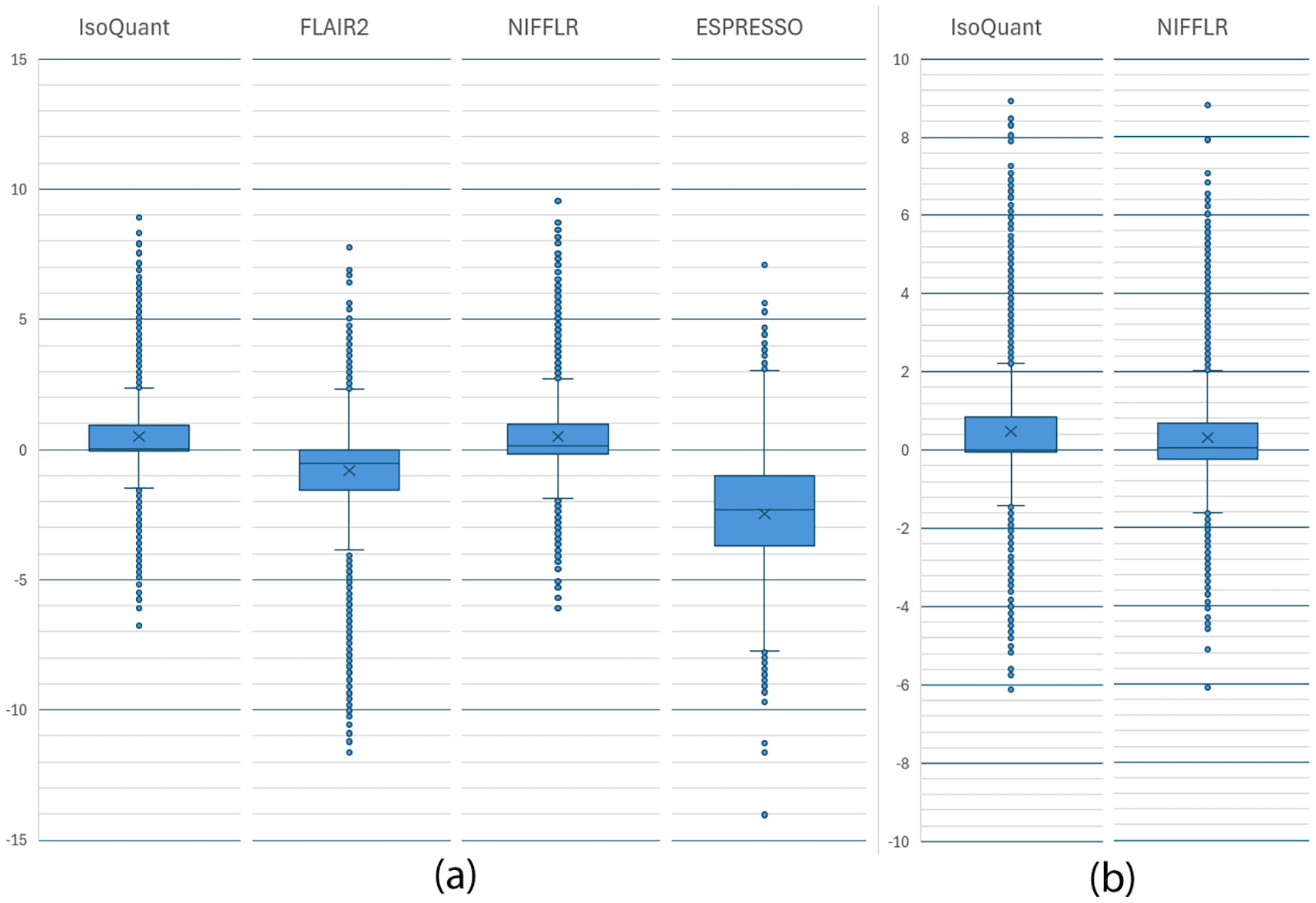
The box spans the upper and lower quartile of the log2 ratios, and the whiskers represent 95% of the values, with individual outliers outside of the 95% interval shown as dots. IsoQuant and NIFFLR show the least variation from the true counts in the simulated data. (b) Box and whisker plots of the log2 ratios of the actual and computed read counts for each transcript from the set of 18,686 simulated transcripts quantified by both NIFFLR and IsoQuant. IsoQuant and NIFFLR show the same accuracy (the height of the box and whiskers are the same size) on this set of transcripts, however, NIFFLR counts have smaller bias (the mean and the median for NIFFLR are closer to zero) and fewer outliers.
For this experiment, we selected the GTEX-1192X sample, which was sequenced with both Illumina and Oxford Nanopore instruments. The ONT data contained 7.6 million long reads with an N50 of 872 bp and a total sequence of 5.3 Gbps. In this dataset, the exact expression of existing and novel transcripts is unknown. However, we can estimate the number and abundances of the known transcripts from the Illumina RNA-seq data, which provides much deeper coverage of the sample. We used StringTie2 (Kovaka et al., 2019) in reference-guided mode to assemble the Illumina data, and this yielded 51,909 distinct transcript variants. The reference-guided mode of StringTie does not output any novel isoforms. Table 2 shows the number of total isoforms and known isoforms found by the four long-read quantification programs when using the ONT data. NIFFLR identified and quantified 43,093 transcripts that matched the reference, which was more than twice as many as any of the other pipelines. To evaluate the accuracy of the quantification, we compared the read counts computed by the programs to the transcript coverage values computed by StringTie on the Illumina data from the same sample. To adjust for the overall coverage difference, we multiplied the coverage values for the Illumina data by 1.59, corresponding to the ratio of the number of bases in the Illumina reads (8.5B bp) divided by the number of bases in the ONT reads (5.33B bp). Figure 4 presents box-and-whisker plots of the ratios (expressed as base-2-logarithms) of the scaled transcript coverages computed with StringTie from Illumina RNA-seq reads and the read counts computed with long-read pipelines from Oxford Nanopore reads for the same sample. The box spans the upper and lower quartile of the ratios and the whiskers represent the range for 95% of the values, with individual outliers outside of the 95% interval shown as dots. The quantification estimates produced by NIFFLR were the second most-consistent to StringTie, outperformed slightly by Isoquant. NIFFLR was the most sensitive, quantifying 26,312 isoforms found in the Illumina RNA-seq data by StringTie.
| # of reference isoforms | # of total isoforms | |
|---|---|---|
| FLAIR2 | 14,957 | 75,557 |
| IsoQuant | 17,183 | 17,183 |
| NIFFLR | 43,093 | 58,377 |
| ESPRESSO | 21,026 | 26,222 |
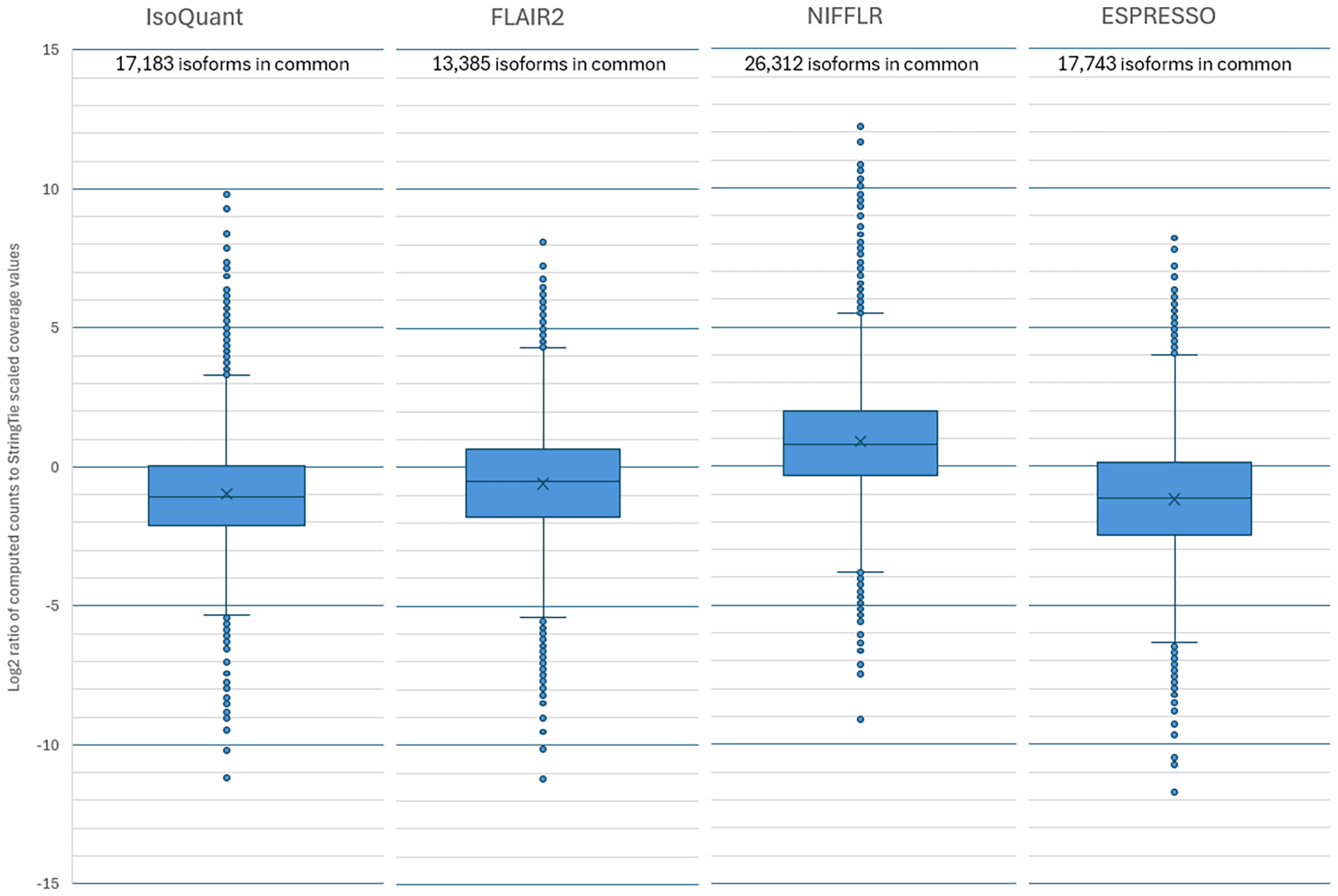
NIFFLR quantified 26,312 reference transcripts that were also quantified with StringTie, far more than the competing pipelines. IsoQuant counts are the most consistent with StringTie counts derived from Illumina data for the same sample, and NIFFLR counts are the second closest.
We applied NIFFLR to identify and quantify isoforms in 92 ONT GTEx samples described in (Glinos et al., 2022), using the RefSeq annotation of GRCh38.p14 as the reference. Across all samples, we identified 135,343 known isoforms and 316,284 novel isoforms in 35,686 genes. Our high confidence set included isoforms identified in two or more sequence samples, and it includes 106,667 novel isoforms and 121,155 known isoforms across 32,875 genes. Number of isoforms identified by NIFFLR far exceeds the number reported by FLAIR (Glinos et al., 2022), which identified 93,718 transcripts across 21,067 genes, of which 77% were novel. Figure 5 illustrates the distribution of counts of novel isoforms across all samples. Interestingly, NIFFLR identified 13 novel isoforms that were present in all 92 samples. Three of these 13 isoforms are annotated in the CHESS annotation version 3.0.1 (Varabyou et al., 2023), or in the GENCODE annotation release 47, with one isoform present in both annotations. Table 3 shows the breakdown of novel and known transcripts found by NIFFLR in GTEx long-read data by tissue. As expected, the percentage of novel isoforms increases with increase in the number of samples for a given tissue, as rare isoforms become more abundant.
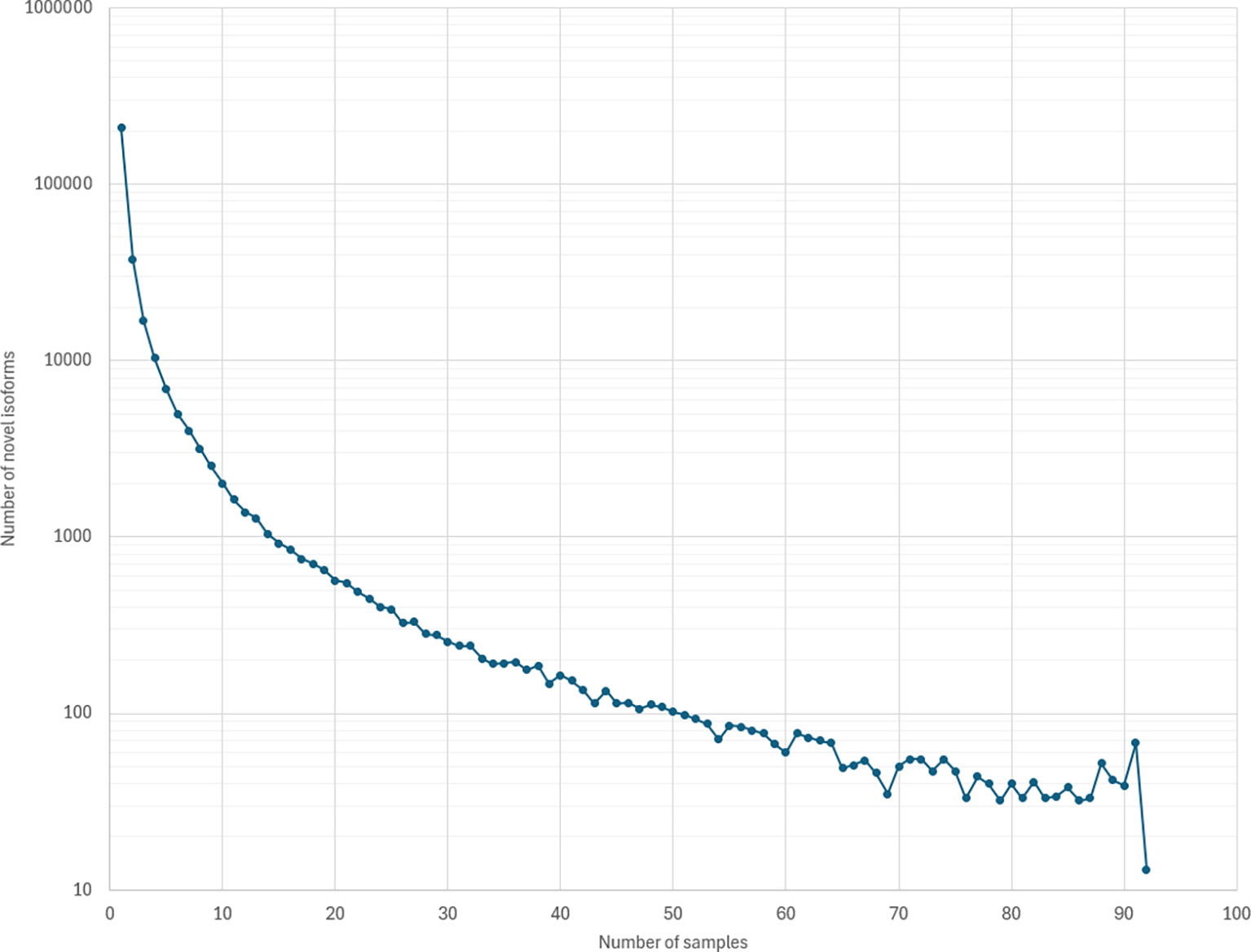
The total number of novel isoforms identified by NIFFLR in the 92 GTEX samples was 451,627. Of these, 223,805 were only seen in a single sample and 13 isoforms were identified in all 92 samples.
The share of novel isoforms increases with the increase in the number of samples for a given tissue. We used all isoforms identified by NIFFLR for the counts shown in this table.
In this manuscript, we describe a novel approach for the discovery and quantification of isoforms from long-read RNA sequencing data produced by Oxford Nanopore sequencing technology. The key difference between NIFFLR and other published programs with similar functionality is that NIFFLR aligns exons from the reference annotation directly to the reads, rather than performing spliced alignment of the reads to the genome. This approach works best for well-annotated genomes, such as the human genome, offering superior sensitivity in this case. However, NIFFLR can still be applied to genomes where their annotation is less reliable, after inferring potential exons from the Illumina RNA-seq data using transcriptome assemblers such as StringTie.
NIFFLR is generally fast enough for research use. As shown in Table 4, NIFFLR was slower than FLAIR2 and IsoQuant, but much faster than ESPRESSO on both simulated and real datasets. Most of the runtime for NIFFLR was spent on aligning exons to the long reads.
We ran all experiments on a 24-core Intel Xeon Gold server with 1TB or RAM, using 24 threads. Time is in hours.
| IsoQuant | FLAIR2 | NIFFLR | ESPRESSO | |
|---|---|---|---|---|
| Simulated reads | 0.7 | 1.3 | 1.9 | 45 |
| GTEx sample | 1.2 | 2.1 | 3.2 | 106 |
NIFFLR is written in shell script, Python, and C++ (the psa_aligner code). To simplify installation, we provide an install script that performs system checks and compiles all necessary executables. We have tested the installation on several popular Linux distributions including RedHat 7, 8, and 9, as well as Ubuntu 18, 20, and 22 LTS.
• Software available from: https://github.com/alguoo314/NIFFLR
• Source code available from: https://github.com/alguoo314/NIFFLR
• Archived source code at time of publication: Zenodo doi 10.5281/zenodo.15585584
• License: GNU General Public License v3.0
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)