Keywords
: Severe haemophilia-A, novel genetic variations, in silico approach, amino acid, molecular technique, intron 22 inversion, intron 1 inversion
This article is included in the Genomics and Genetics gateway.
: Severe haemophilia-A, novel genetic variations, in silico approach, amino acid, molecular technique, intron 22 inversion, intron 1 inversion
Haemophilia-A is a coagulation disorder caused by a deficiency of factor VIII (FVIII). It is the most common type of haemophilia in the world, with 50% of the patients having the severe type.1,2 According to World Federation of Haemophilia (WFH) in 2021 and based on Indonesia Haemophilia Society (HMHI), 185,318 and 2,424 patients are clinically diagnosed with Haemophilia A.3,4 This disorder occurs due to genetic variations in FVIII gene that causes a total or partial deficiency of FVIII or dysfunctional FVIII gene. Intron 22 inversion is the most common genetic mutation for severe haemophilia A. In this condition, the FVIII production does not occur, resulting in congenital absence of FVIII. As a result, these patients are susceptible to bleeding, especially joint and muscle bleeding. Generally, these patients experience bleeding not just in joints, but also in muscles, soft tissues, or internal organs with or without trauma which can occur since the first year of life.1,5,6 Recurrent joint bleeding can cause persistent joint damage or haemophilia arthropathy and reduce the patient’s quality of life. In addition, spontaneous bleeding in internal organs can cause death.5–8 Genetic variations that result in the absence of FVIII production, resulting in failure to form immune tolerance to FVIII, are associated with a high risk of inhibitors.9,10
Currently there is no data on genetic variations that occur in haemophilia-A patients in Indonesia nor the molecular technique to observe the genetic variations within the FVIII gene. The diagnosis of haemophilia-A is established based on FVIII level examination without identifying the genetic variation that occurs. This study aims to develop and modify a molecular technique to detect intron 22 and intron 1 inversion among hemophilia patients in Indonesia, observe any novel genetic variations, and classifying them according to American College of Medical Genetics and Genomics (ACMG 2015, with current updates on CanVIG-UK) guidelines. Knowledge of the genetic variations will be useful for confirming the diagnosis of haemophilia-A, knowing the pattern of bleeding and joint damage as well as the risk of FVIII inhibitors, and guiding the administration of clotting factor concentrates. The genetic mutation information is also useful in genetic counselling for families.11 Therefore, this concludes the novelty from this research.
Ten (10) participants recruited for this study are haemophilia-A patients that registered in Dr. Cipto Mangunkusumo Hospital, DKI Jakarta, Indonesia. This pilot study was conducted after obtaining informed consent from patients and their parents. The informed consent was written and signed by the participant. This study was approved by the Ethics Committee of the Faculty of Medicine, Universitas Indonesia (No: KET-490/UN2.F1/ETIK/PPM.00.02/2024 with protocol number: 24-02-0332). Three ml of blood sample were collected in EDTA tubes and stored at 4 °C until genomic DNA (gDNA) extraction was performed.
As much as 555 μl raw samples were put into a 1.5 ml microtube. A volume of 1000 μl of RBC lysis buffer reagent (Geneaid Biotech Ltd, Taiwan)12 was added to the sample. The samples were incubated for 18 minutes. The microtubes were then centrifuged (Eppendorf 5424R, Germany) at 3,000 g for 5 minutes. A white precipitation formed at the bottom of the microtubes. Supernatant then were discarded. A volume of 100 μl RBC lysis buffer (Geneaid Biotech Ltd, Taiwan) and 200 μl GB buffer were added (Geneaid Biotech Ltd, Taiwan) and mixed thoroughly by shaking the microtubes. The samples were incubated for 11 minutes in 65 °C heater block (Stuart SBH130D, England) and then transferred to a new sterile GD Column. Samples then were centrifuged in an Eppendorf 5424R, Germany for 5 minutes with 15,000 x g (room temperature) and the supernatant was transferred to a fresh, sterilized microcentrifuge tube. A volume of 400 μl W1 buffer (Geneaid Biotech Ltd, Taiwan) was added. The samples were then centrifuged in an Eppendorf 5415R at 15,000 x g (room temperature) for 1 minute. A volume of 600 μl wash buffer (Geneaid Biotech Ltd, Taiwan) was added. The samples were then centrifuged (Eppendorf 5415R) at 15,000 x g (room temperature) again for 1 minute. After replacing the GD column into a fresh new collection tube, 100 μl of elution buffer (Geneaid Biotech Ltd, Taiwan) was added carefully, and the sample was incubated for 3 minutes. After 3 minutes of incubation, the tubes were centrifuged (Eppendorf 5415R) for 1 minute with 15,000 x g (room temperature). The genomic DNA (gDNA) samples were frozen at −20 °C for further use.
Genomic DNA concentration and purity were quantitatively assessed using a spectrophotometric assay in Varioskan microplate reader (Thermo Fisher Scientific; United States).13 Absorbance was measured at wavelengths of 260 and 280 (A260 and A280, respectively) nm. The absorbance quotient (OD260/OD280) was used to estimate DNA purity. An absorbance quotient ratio between 1.8 and 2.0 was considered good for purified gDNA. A ratio < 1.8 indicated protein contamination, while a ratio of >2.0 indicated RNA contamination.
As much as 15 μl of gDNA then were digested using 1.5 μl of BCl1 enzyme in 28.5 μl Nuclease Free Water (NFW) + 15 μl buffer. The digested products were incubated in 55.3 °C overnight. Samples then went to purification using column with addition of 116 μl TE buffer. As much as 116 μl products from previous steps, a 1 μl volume of T4 ligation enzyme +13 μl buffer were added. Samples were then incubated in 15 °C overnight. The ligated products then went to purification using column with addition of 36 μl TE buffer. This final product then was storage in -25 °C for further use.
The gene used in this study is the FVIII gene (NM_000132.4), located in chromosome Xq28, consisting of 26 exons. FASTA sequence of the gene was obtained from NCBI (https://www.ncbi.nlm.nih.gov/nuccore/NG_011403.2?report=fasta&from=5001&to=191932) while the FASTA coding sequence (CCDS) was obtained from Ensembl! (https://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi? REQUEST = CCDS&DATA = CCDS35457). All FASTA sequence was annotated using Benchling (https://www.benchling.com/), while primer for polymerase chain reaction (PCR) was designed and the quality were assessed using NetPrimer (http://www.premierbiosoft.com/NetPrimer/AnalysePrimer.jsp) and NCBI Primer Blast (https://www.ncbi.nlm.nih.gov/tools/primer-blast/).
The IS-PCR for the intron 22 inversion and intron 1 inversion of FVIII gene was performed using a multiplex primer pair and its sequences as mentioned in Table 1. Primer sequence for exon 14 Sanger sequencing is also mentions in Table 1. MyTaq HS Redmix (Meridian Biosciences; United States)14 was used as a PCR mix for both PCR products. The PCR program for IS-PCR and gradient PCR consisted of pre-denaturation at 95 °C for 1 min, denaturation at 95 °C for 15 sec, annealing at 65 °C for 15 sec, extension at 72 °C for 10 sec, repeated for 38x cycles and completed by a final extension at 72 °C for 10 min. Agarose gel electrophoresis was performed to verify the quality of the PCR products. The PCR products were separated on a 1.5% agarose gel at 100 volts for 45 minutes. The gels were visualized and analysed using a gel documentation system (Accuris Instruments; United States).15 PCR products for exon 14 then were sequenced using sequencer and the analysis were performed using FinchTV and BioEdit.
Result obtained from the IS-PCR are then to be analyzed visually using the gel documentation system (Accuris Instruments; United States).14 The amplicon length measured using the 50 basepair (bp) DNA ladder under the blue light inside the chamber. Photos then were taken using smartphone camera for further interpretation.
All identified novel variants in the FVIII gene were classified according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines, as described by Richards, et al. (2015).16 Variant interpretation followed a semi-quantitative framework incorporating population data, computational predictions, functional studies, segregation data, and published literature. Each variant was evaluated against a set of criteria defined by the ACMG/AMP, encompassing pathogenic (P), likely pathogenic (LP), variant of uncertain significance (VUS), likely benign (LB), and benign (B) categories. Population frequency was assessed using databases such as gnomAD and ExAC. In silico predictive tools, including PolyPhen-2, SIFT, and CADD, were used to assess the potential impact of missense variants.
Functional evidence, when available from in vitro or in vivo studies, was incorporated under the PS3 or BS3 criteria. Variants previously reported in association with hemophilia A were reviewed from databases such as HGMD, ClinVar, and LOVD, and evaluated under the PS1, PS4, and PP5 criteria. Co-segregation with disease within families, de novo occurrence, and phenotypic concordance were evaluated where relevant and when data were available. For null variants (nonsense, canonical splice site, and frameshift), classification considered whether loss-of-function is a known mechanism of disease in FVIII (PVS1). All classifications were independently reviewed by at least two trained analysts, and discordances were resolved by consensus discussion or consultation with a clinical geneticist.
Sequencing result in FASTQ format obtained was analyzed using FinchTV version 1.4 (downloaded from https://finchtv.software.informer.com/1.4/#google_vignette) and BioEdit version 7.2 (downloaded from https://bioedit.software.informer.com/7.2/) software to align its genes. Wildtype gene FASTA sequence was obtained from NCBI (https://www.ncbi.nlm.nih.gov/nuccore/NG_011403.2?from=5001&to=191932&report=fasta) and wildtype coding sequence (CCDS) was obtained from Ensembl! (https://asia.ensembl.org/Homo_sapiens/Psychic?site=ensembl&q=FVIII). Computational and predictive data were obtained by the clinically significant of each variant, and were assessed using the consensus scoring of Varsome (https://varsome.com/). The classification will be divided into pathogenic, likely pathogenic, uncertain significance (VUS), likely benign, and benign. Each variants effect on amino acid interactions, bonding, and side chain were all analyzed using Biovia discovery studio 24 (downloaded from https://discover.3ds.com/discovery-studio-visualizer-download ). Wildtype amino acid sequence in PDB format was obtained from accessing the Alphafold protein structure database (https://alphafold.ebi.ac.uk/entry/P00451). The wildtype sequence of the FVIII gene will be imported to Biovia discovery studio 24 and annotated the mutation point on amino acid sequence to analyse the change in its interactions.
Result obtained from this research shown that there is different size in amplicon which determined the intron 22 and intron 1inversion, and negative control. As depicted in Figure 1, the schematic primer design is meant to easily distinguish between patients and negative control. The IS-PCR is based on the principle of restriction enzyme digestion of genomic DNA followed by self-ligation, resulting in circular DNA fragments. These fragments contain the inversion breakpoints in rearranged orientations that are distinguishable by specific primer pairs. PCR amplification across the ligated junctions allows the detection of both normal and inverted configurations. For intron 22 inversion, the IS-PCR exploits the presence of homologous intragenic and extragenic repeats which mediate non-allelic homologous recombination. Similarly, intron 1 inversion is caused by recombination between homologous sequences located within intron 1 and upstream of the FVIII gene. The IS-PCR assay yields distinct amplification patterns for normal and inverted alleles, enabling precise molecular diagnosis of haemophilia-A caused by these recurrent rearrangements.17 As this method was conducted to be the first step in screening for molecular diagnosis in haemophilia A patients, we tried to conduct a simpler way to analyse its differences in-between patients.
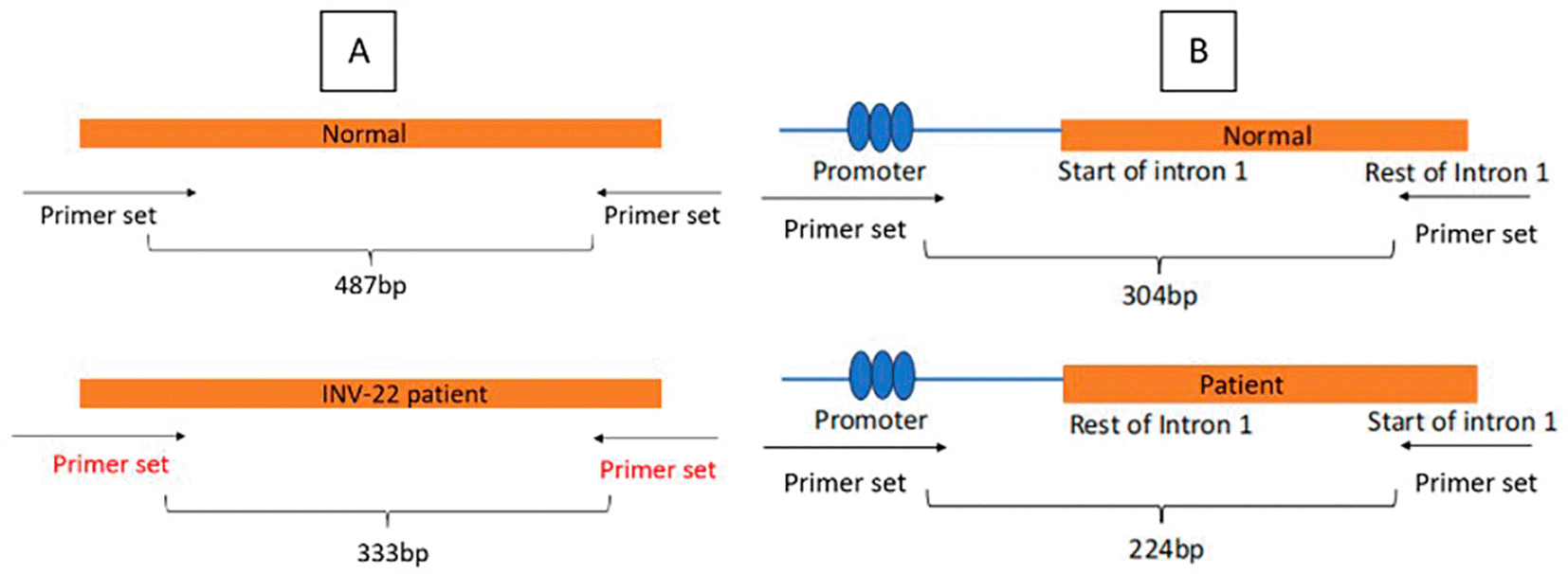
All gDNA extracted, digested, and ligated were in good conditions. The concentration exceeded the recommended level and the purity is within range. The IS-PCR shown that the schematic primer works and shows different size in amplicon. Figure 2 mentioned below shows the final result of IS-PCR from intron 22 and intron 1 inversion patients and negative control. Subject with intron 22 inversion showed 333 basepair in its amplicon size, while negative control showed 487 basepair. Subject with intron 1 inversion showed 224 basepair in its amplicon size, while negative control showed 304 basepair. This method has also been discovered by Rosetti et al. (2008)17 whose results aligned with this study. We accomplish a result with clear amplicon bands of the expected size for the corresponding PCR products as depicted in Figure 2.
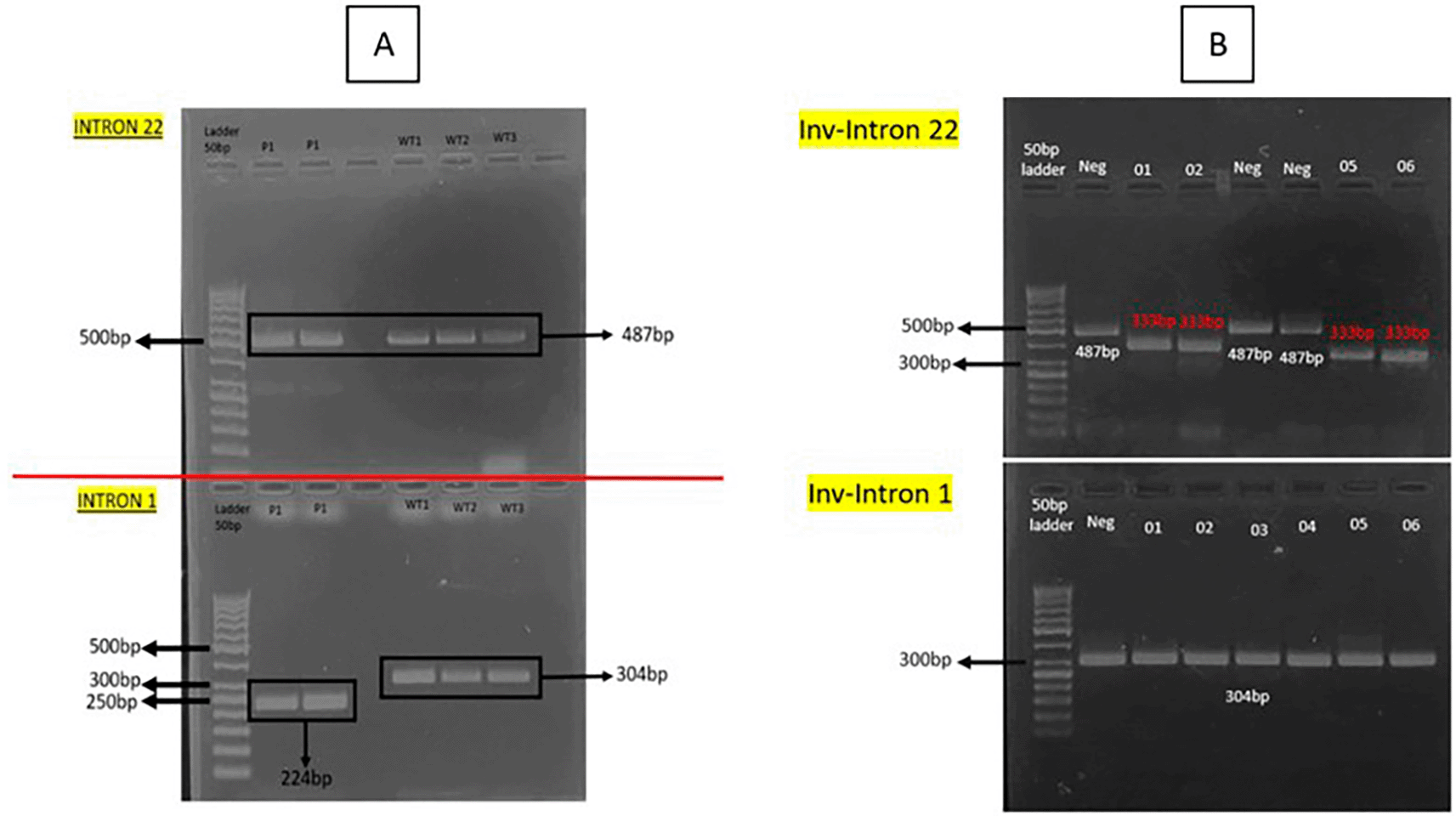
In addition to being highly reproducible, our genotyping results showed the sizes expected for wildtype and inversions patients with minimal variability among different time of research. These trivial discrepancies can be ascribed to differences in the running temperature, gel polymer, or running buffers. Noticeably, our results were consistent and reproducible with different amounts of ligation product in the PCR reaction and in different experiments with the same sample. The original IS-PCR protocol of Rossetti et al. (2008),18 designed with basic methods of molecular biology, can be used worldwide as a screening program to detect the most common pathogenic variants in the haemophilia population. Our modification within this protocol requires specialized infrastructure, which is generally available in research center or universities, but is mostly unavailable in the clinical facilities of public health institutions. This problem is also in accordance with research from Martinez-Contreras et al. (2024).19 However, Considering the advantages of sensitivity and reliability, our improved method may be implemented in regional center of reference to diagnose haemophilia A and other rare diseases in Indonesia.
According to the result of Sanger sequencing, it is shown that there are nucleotide changes in exon 14 of FVIII gene which are annotated in Benchling. This novel variant showed changes in coding sequence (CDS) position 2871, 3637, and 3640. As a consequence of nucleotide changes, amino acid changes were also observed in position 957, 1213, and 1214. All three novel variants are annotated as c.2871G > C; p.(Leu957Phe), c.3637A > T; p.(Ile1213Phe), and c.3640; p.(Gln1214Ter) as depicted in Figure 3A, 3B, and 3C respectively. The classification of all novel variants observed in this study is according to ACMG-AMP.16
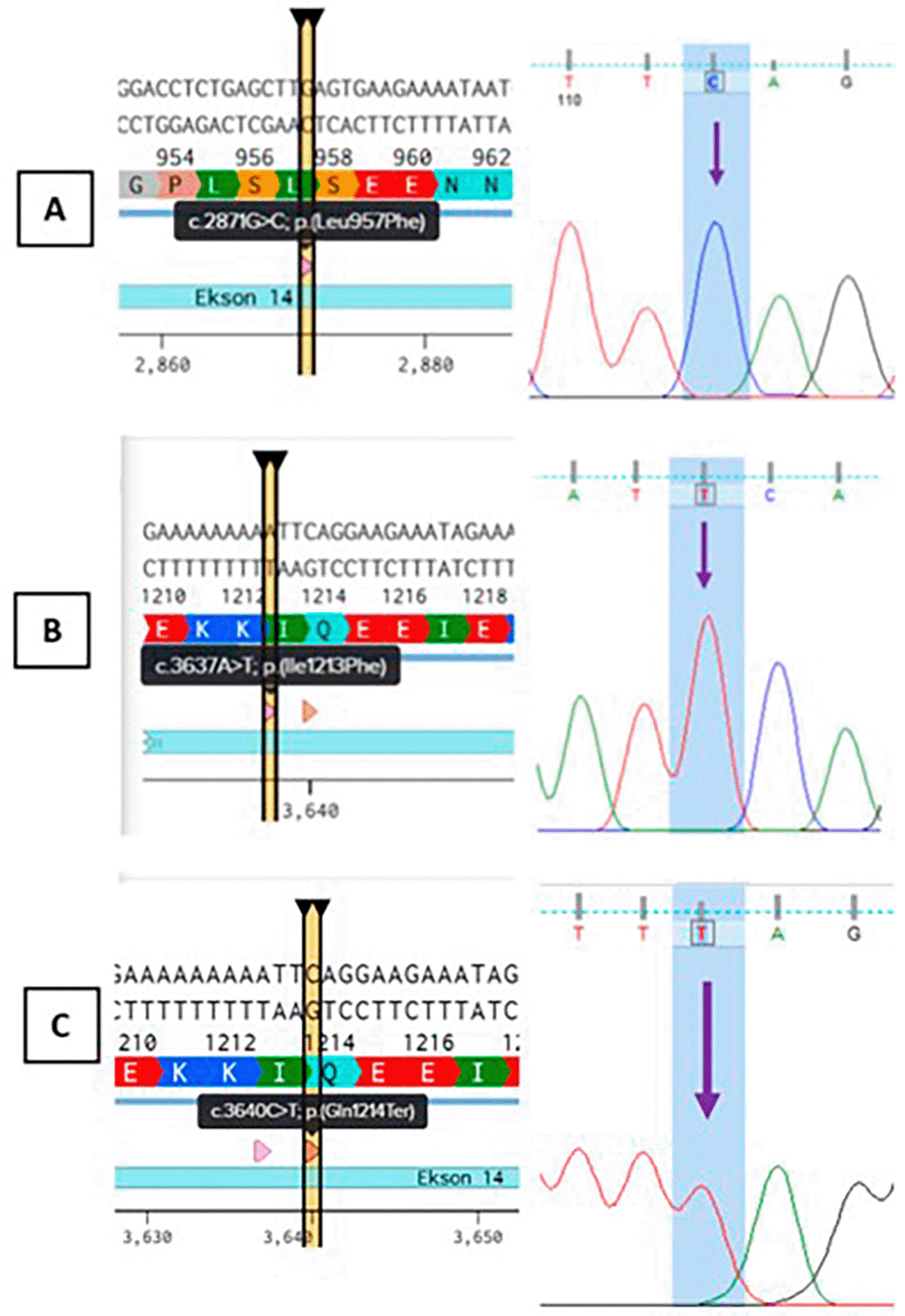
According to Richards et al. (2015),16 this variant is absent in normal population database when checked using gnomAD v4.1.0 GRCh 38.20 This evidence met the criteria for population data of PM2_supporting. Computational and predictive data shown that the two missense novel variants (c. 2871G > C; p.(Leu957Phe) and c.3637A > T; p.(Ile1213Phe)) are predicted to have deleterious effect on gene or gene product, which met the criteria for PP3. The nonsense variant c.3640C > T; p.(Gln1214Ter) is predicted to undergo nonsense-mediated mRNA decay (NMD). NMD is a cellular mechanism that degrades mRNA transcripts containing premature stop codons, preventing the production of truncated proteins that may be deleterious. According to Abou-Tayoun et al (2018)., if the nonsense variant is predicted to undergo the NMD mechanism and the exon is proven to be present in biologically-relevant transcript, this met the criteria for PVS1. This declared that the predicted nonsense variant in gene with loss of function (LoF) is a known mechanism of disease.21 As this variant leads to early termination and degradation of the transcript, it results in no functional factor VIII protein. Based on clinical examination, all subjects have FVIII less than normal and diagnosed with haemophilia A. According to phenotype to genes database, the FVIII protein is encoded only by FVIII genes. This makes the phenotype observed is unique and highly specific to gene etiology. As the “other data” section is being updated by Basel-Salmon, L. (2024), this phenotype met the criteria for PP4_strong.22 All evidence collected to classify the three novel variants are summed up in Table 2.
The protein structure of FVIII gene is depicted in Figure 4. The 3D protein model has a high confidence. The full length of amino acid (AA) translation consisted of 2,351 AA. FVIII is a large glycoprotein involved in the intrinsic pathway of blood coagulation, functioning as a cofactor for factor IX-A to activate factor X. The full-length FVIII protein consists of 2,351 AA and is composed of the domain structure A1–A2–B–A3–C1–C2. As summarized in Table 3, the A1, A2, and A3 domains is homologous to ceruloplasmin, important for cofactor activity and binding to factor IX-A. The B domain is heavily glycosylated, non-essential domain that is removed during activation. The C1 and C2 domains are important for phospholipid binding and interaction with von Willebrand factor (vWF), which stabilizes FVIII in circulation.16,23 A full-length protein will also have normal function whilst a mutation in gene will lead to disease-causing. The AlphaFold-predicted structure of FVIII protein helps visualize which parts of the protein are structurally robust or flexible. Mutations occurring in high-confidence structured domains (A and C domains) are often more damaging and correlate with severe haemophilia A phenotypes. Structural data integrated with variant interpretation can significantly enhance the clinical classification of FVIII genetic variations.
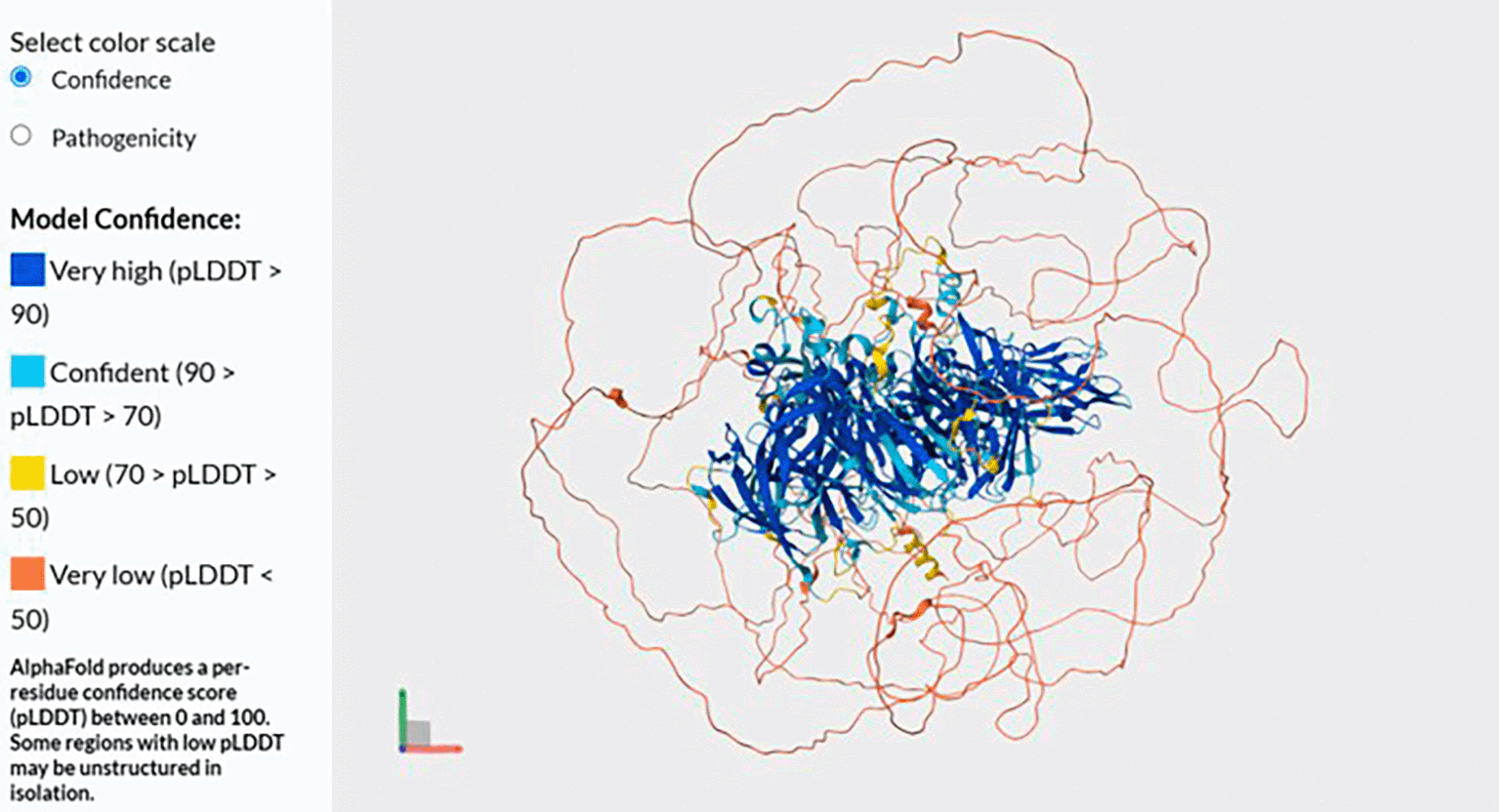
Source: Uniprot (https://www.uniprot.org/uniprotkb/P00451/entry#structure)
The first missense variant found in this study (c.2871G > C; p.(Leu957Phe) leads to AA change and affect their interaction. According to Figure 5, a variant occuring in c.2871 with nucleotide changes from Guanine (G) to Cytosine (C) leads to AA change in position 957. The AA change from Leucine (Leu) to Phenylalanine (Phe) is predicted to disrupt the function of FVIII protein. According to analysis, the substitution of leucine to phenylalanine at position 957 (Leu957Phe) in the FVIII protein involves replacing a small, aliphatic, nonpolar residue with a bulkier, more rigid aromatic amino acid. Leucine normally contributes to the hydrophobic core of the A2 domain through flexible van der Waals interactions that support the protein’s structural stability. In contrast, phenylalanine introduces a phenyl ring that is more rigid and significantly larger. This change can disrupt the local packing environment, potentially causing steric clashes or destabilization of the domain’s three-dimensional structure.25
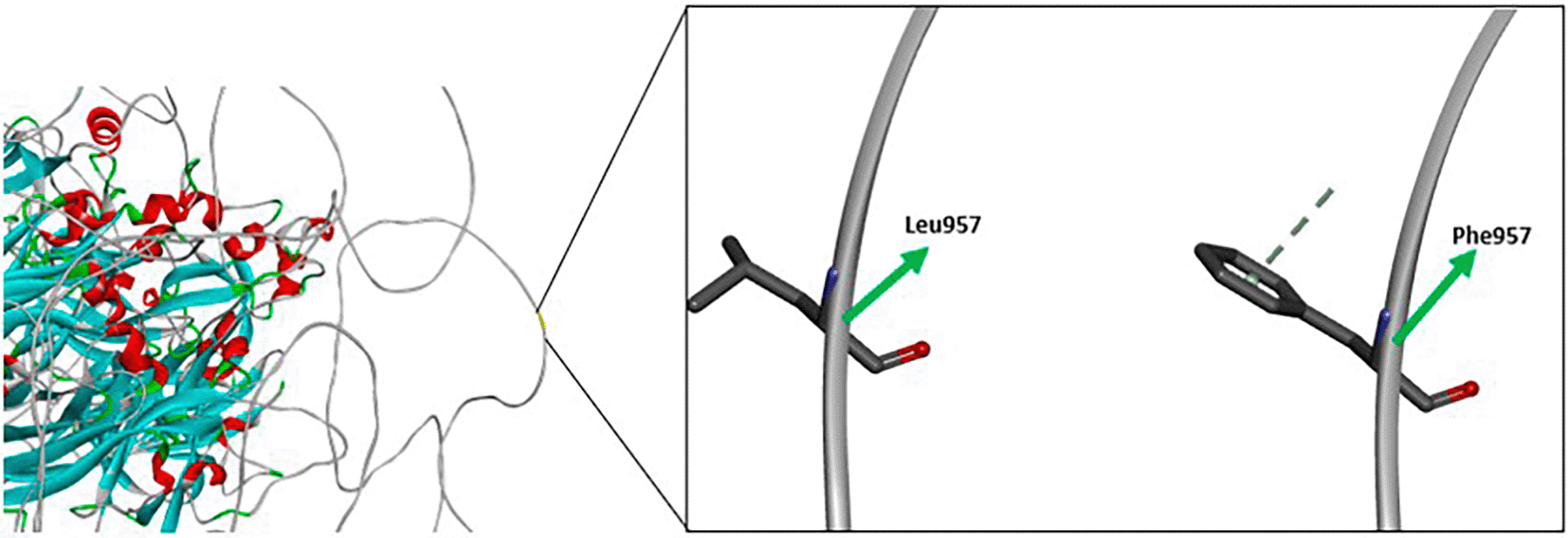
As a result, the substitution may impair proper folding of the A2 domain, leading to reduced secretion of functional FVIII protein or degradation in the endoplasmic reticulum. Since the A2 domain is crucial for FVIII’s interaction with activated Factor IX (FIXA) during the coagulation cascade, structural disturbances here can lead to impaired cofactor activity. Clinically, such a mutation is often associated with moderate to severe haemophilia-A, depending on how much functional FVIII protein remains.25,26
Variant occurring in c.3637, the nucleotide changes from Adenine (A) to Thymine (T), leading to AA changes in position 1213. This variant resulted in AA changes from Isoleucine to Phenylalanine. The image depicted in Figure 6 illustrates a structural comparison of the amino acid substitution Ile1213Phe in the FVIII protein. The wild-type residue isoleucine (Ile) at position 1213 is shown with a compact, branched aliphatic side chain. This hydrophobic, flexible side chain plays a role in maintaining the protein’s core packing and local structural stability. On the right, the substituted residue phenylalanine (Phe) is depicted with a much bulkier, rigid aromatic ring.27
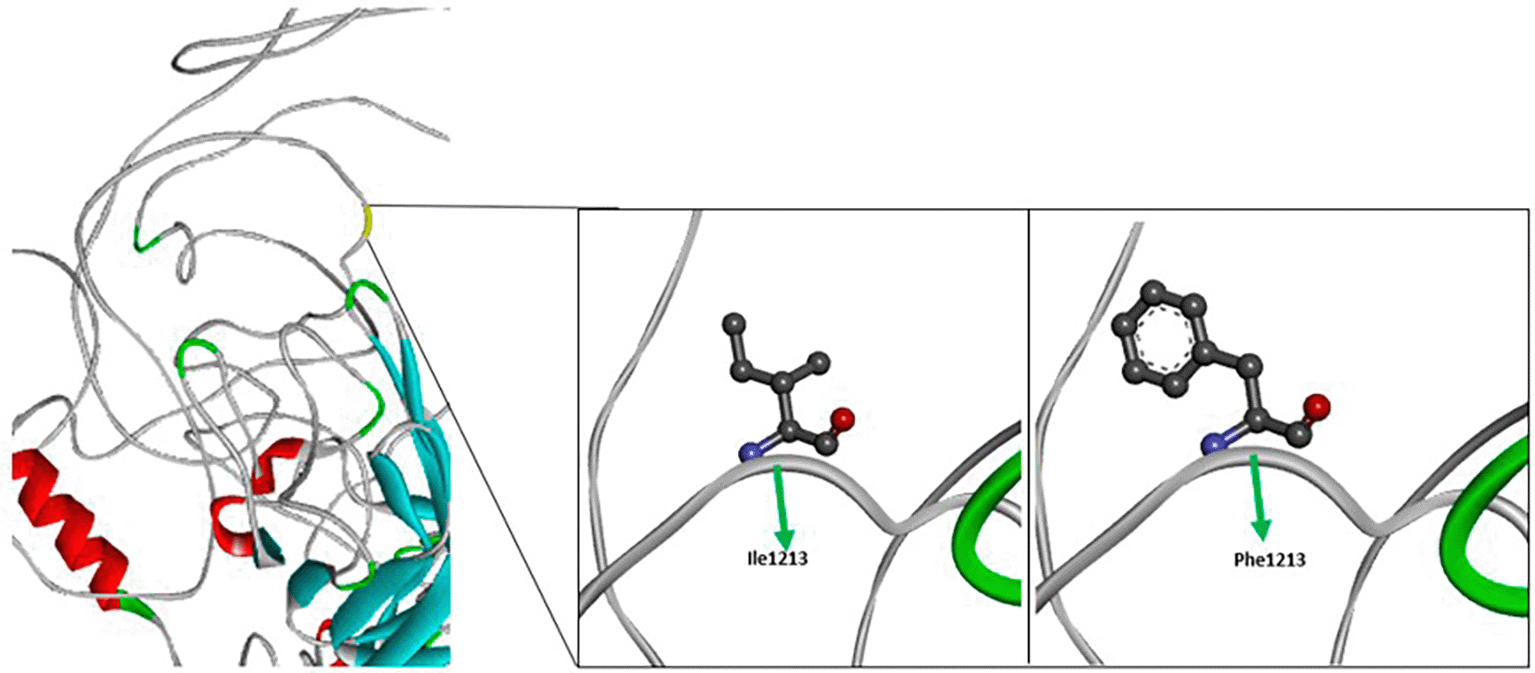
While both residues are nonpolar, the replacement introduces a phenyl group that can significantly alter the local environment due to its larger size and potential for π-π stacking or steric interactions. This change from Ile to Phe at position 1213 occurs in the A3 domain of FVIII, which contributes to the interaction with vWF and stabilization of FVIII in circulation. The presence of a bulkier aromatic side chain at this position may interfere with the proper folding of the domain or disrupt essential hydrophobic interactions, leading to decreased protein stability, impaired interaction with VWF, or reduced FVIII secretion. These structural changes can compromise FVIII function in the coagulation cascade, potentially contributing to haemophilia-A of variable severity depending on the residual activity of the mutated protein.23,27
One novel nonsense variant in exon 14 of FVIII gene which is annotated as c.3640C > T; p.(Gln1214Ter) results in a nonsense mutation at codon 1214, replacing an Glutamine (Gln) with a premature stop codon (Ter), denoted as p.(Gln1214Ter). This mutation occurs in exon 14, within the A3 domain of the Factor VIII (FVIII) protein, which is essential for stabilizing FVIII and mediating its interaction with vWF in plasma. The presence of a premature termination codon (PTC) at position 1214 leads to two likely consequences. First, it may trigger nonsense-mediated mRNA decay (NMD), a cellular surveillance mechanism that degrades transcripts containing early stop codons to prevent production of truncated, potentially harmful proteins. Given that this PTC is located well upstream of the last exon-exon junction, the transcript is predicted to be a target for NMD, therefore substantially reducing or eliminating FVIII protein synthesis.23
If NMD does not occur, the truncated FVIII protein would lack the C1 and C2 domains, which are critical for membrane binding and interaction with VWF. Such a truncated protein would be non-functional, as it cannot anchor to phospholipid surfaces or remain stable in circulation. This strengthens the prediction that frameshift variants greatly influence the overall protein structure, interactions, and bonds. Apart from that, the subjects’ clinical condition also in accordance with all the variants found in this study.23,28,29
This study successfully conducted modified technique to detect genetic variation in the FVIII gene. Using IS-PCR, intron 22 and intron 1 inversions were clearly detected through distinct amplicon patterns, confirming the utility and reproducibility of this method for routine molecular diagnosis. Additionally, three novel variants in exon 14—c.2871G > C (p.Leu957Phe), c.3637A > T (p.Ile1213Phe), and c.3640C > T (p.Gln1214Ter)—were identified via Sanger sequencing and classified using ACMG/AMP guidelines. Structural and computational analyses showed that the missense variants (Leu957Phe and Ile1213Phe) disrupt local hydrophobic interactions and likely impair folding and stability of the A2 and A3 domains, respectively. Meanwhile, the nonsense variant (Gln1214Ter) introduces a premature stop codon predicted to undergo nonsense-mediated mRNA decay, abolishing FVIII protein production. All variants correlated with clinical severity (FVIII level less than normal), supporting their pathogenic classification and underscoring the importance of comprehensive molecular diagnostics in managing haemophilia A in the Indonesian population.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)