Keywords
workflow, pipeline, graph, WDL, Nextflow, BioUML
This article is included in the Bioinformatics gateway.
workflow, pipeline, graph, WDL, Nextflow, BioUML
Workflow languages are widely used when production systems analyze biomedical data since they provide consistent processing of batches of data, reproducible processing of a particular dataset, restart and reentry, data provenance, and other desirable properties, such as the ability to easily share bioinformatics pipelines with others.1
For this reason, a wide variety of workflow languages and workflow managers are used in bioinformatics. For example, the most complete list of computational data analysis workflow systems2 contains 350+ entries.
The popularity of particular workflow languages and workflow managers varies by subfield, data type, computing environment, and over time. Table 1 summarizes the number of workflows by used languages in the main workflow registries related with bioinformatics: Dockstore3 and WorkflowHub.4
WDL - Workflow Description Language, CWL - Common Workflow Language.
| Workflow language | Dockstore | WorkflowHub |
|---|---|---|
| WDL | 4603 | 8 |
| CWL | 232 | 111 |
| Nextflow | 201 | 167 |
| Galaxy | 159 | 713 |
| Other | - | 352 |
Here we briefly describe the top 4 workflow languages listed in Table 1 and corresponding workflow systems.
WDL - Workflow Description Language5 was originally developed for the Broad Institute’s genomic analysis pipelines. Now it is an open standard for describing data processing workflows with a human-readable and writeable syntax. WDL makes it straightforward to define analysis tasks, connect them together in workflows, and parallelize their execution. The language enables common patterns, such as scatter-gather and conditional execution, to be expressed simply.6
To execute WDL scripts there are two main workflow engines:
- Cromwell7 - is a production-grade workflow execution engine developed by the Broad Institute. It is written in Scala and is designed for high-scale execution of WDL workflows, particularly in cloud environments like Google Cloud Platform (GCP) or on local clusters. Many users run their WDL workflows in Terra,8 a managed cloud bioinformatics platform with built-in WDL support provided by Cromwell.
- miniWDL9 - is a lightweight WDL runtime and developer toolkit, developed by the Chan Zuckerberg Initiative. It is written in Python and is primarily geared towards local development, testing, and prototyping of WDL workflows. It can execute WDLs, and is often favored for its ease of installation and use in a local development environment, providing tools for linting, parsing, and running WDLs with minimal setup compared to Cromwell.
CWL - Common Workflow Language10,11 is another open, community-led standard designed for describing analysis workflows and command-line tools in a portable and scalable way. CWL workflows use YAML syntax to define inputs, outputs, and execution steps, allowing them to be run consistently across different computing environments, including workstations, clusters and clouds. cwltool12 is the reference implementation of the CWL standards. Other implementations are listed at the dedicated website.13
Nextflow14,15 is an open-source workflow management system and a domain-specific language (DSL) designed for writing and executing scalable and reproducible scientific workflows. It is based on a reactive dataflow programming model, where tasks are triggered automatically when input data becomes available or changes, allowing natural parallelism without the user having to manage low-level details of task scheduling or resource locking. The DSL, based on Apache Groovy, lets users define workflows that integrate scripts in multiple languages (bash, Python, R, Ruby, etc.) and can reuse existing software tools without wrappers.
nf-core16,17 is a community-driven project that provides a curated set of standardized, best-practice analysis pipelines built using Nextflow. Significant effort has gone into developing tools, templates, and guidelines that enable domain experts to contribute to the community. The result is a set of high-quality pipelines that are portable, reproducible, fully documented, and cloud-ready.
Seqera18 is the company behind Nextflow and has supported the nf-core community since its inception. It has also developed the commercial workflow system Sequera platform and a powerful AI tool specializing in bioinformatics and nextflow language in particular - Sequera AI.19
Galaxy20,21 is an open-source, web-based bioinformatics workflow system implemented using the Python programming language. It is designed for accessible, reproducible, and transparent computational research. Its key feature is graphical workflow creation. Galaxy makes thousands of third-party open-source analysis packages easy to use, and interoperable without any user supplied code. For any new analysis package to become a tool, a developer prepares a Galaxy wrapper once, and uploads it to the sharable Galaxy global tool ‘appstore’ called the Galaxy Toolshed.22 However it uses proprietary JSON format to describe workflow components.
It is desirable to have reliable tools that translate a workflow from one workflow language to another. There are several projects that try to solve this task.
Seqera AI is a commercial tool that is able to convert CWL or WDL to Nextflow and vice-versa using Claude LLM. Seqera AI provides translation with step by step explanation and analysis of the script. It is also able to run generated Nextflow, find errors and automatically fix it. Seqera AI is a great tool for generating and modifying pipelines, studying Nextflow and running it with uploaded input files. However as an LLM it has certain limitations. It works without a clear algorithm of pipeline generation. Sometimes generated workflows are overcomplicated. Sometimes LLM hallucinates and produces scripts with errors which can not be easily traced. It also can get into never ending self fixing cycles. According to information at https://seqera.io/user may use Seqera AI freely with 250 chat sessions per month. Generation of even the simplest pipeline takes some time as requests should be passed to LLM.
CNT1 is a semiautomated translator converting CWL workflows into Nextflow ones. It integrates tool-level conversion, graph dependency analysis, and correctness checks to provide highly automated translation coverage, significantly reducing the development time while satisfying language-specific requirements like building a proper dataflow model when creating workflows. According to the authors estimate it can cover up to 81% of the original workflows.1
Janis23 is a framework creating specialised, simple workflow definitions that are then transpiled to CWL or WDL. Conversion to Nextflow is currently under progress.
cwl2nxf24 is a prototype conversion tool from CWL to Nextflow. However it is abandoned now.
Since 2001 we have been developing the BioUML25 platform - an integrated environment for systems biology and collaborative analysis of biomedical data. Like the Galaxy platform it provides a graphical web interface for workflow visualisation, creation and editing. geneXplain platform is a commercial version of BioUML platform and provides thousands of ready workflows for analysis of different omics data.
BioUML uses its own format for workflow storage and engine for workflow execution. BioUML is also integrated with Galaxy, cwlTool, cromwell and Nextflow for execution of workflows in Galaxy, CWL and Nextflow formats correspondingly. However this integration is quite difficult and is not seamless.
So now we are designing a new architecture where workflows in different formats can be converted into a common object-oriented model presented by a set of Java classes. This model can be visualised and edited as a graph. Then workflow text in different formats can be generated from this model.
As a first step of this project we describe here mapping of WDL workflows into a common workflow model, editing graphs visually and generation of WDL and Nextflow scripts from this model.
We have split corresponding parts of code from the BioUML project into an independent project on GitHub: https://github.com/genespace-ru/workflow-engine . We hope that the created command line application, web application and web service will be useful for many users and can be used as parts of other projects related to workflows.
BioUML meta model. Our tool is created on the base of the BioUML software (www.biouml.org). The meta-model is the core of the BioUML platform.26 It provides an abstract layer (compartmentalized attributed graph) for comprehensive formal description of a wide range of biological and other complex systems. The content of databases on biological pathways (for example Reactome, PantherDB), SBML models, biological pathways in BioPAX format as well as workflows can be expressed in terms of the meta-model. This formal description can be used both for visual representation of the biological system structure and for automated code generation to simulate the model behavior.
The meta-model describes the system as three interconnected parts:
1) graph structure - the system structure is described as compartmentalized graph;
2) database level - each graph element can contain a reference to an object in some database;
3) executable model - any graph element can be associated with an element of a domain-specific model e.g. mathematical model or workflow.
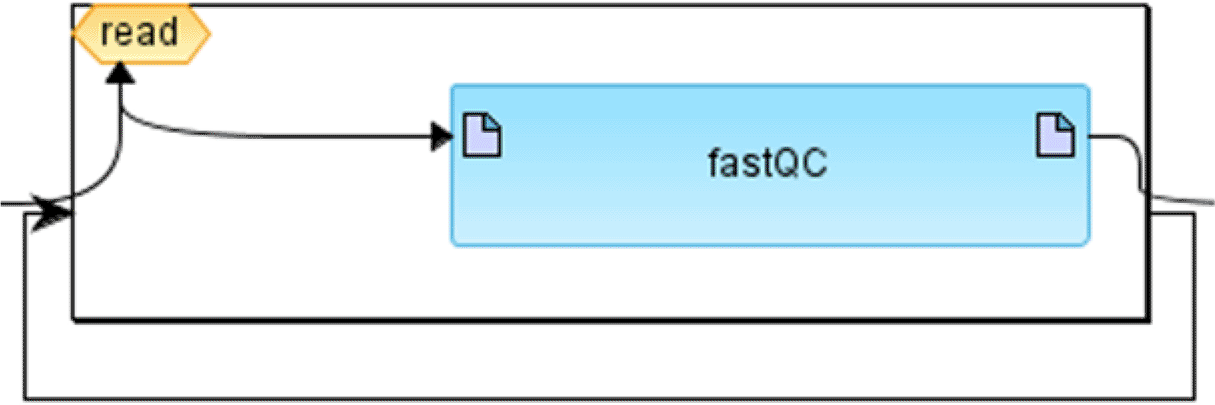
It should be noted that the structure of the meta-model itself is problem-domain neutral and can be applied to a variety of fields including biological models or executable workflows for biomedical data analysis. Figure 1 demonstrates an example of how WDL workflow can be presented in terms of this meta-model.
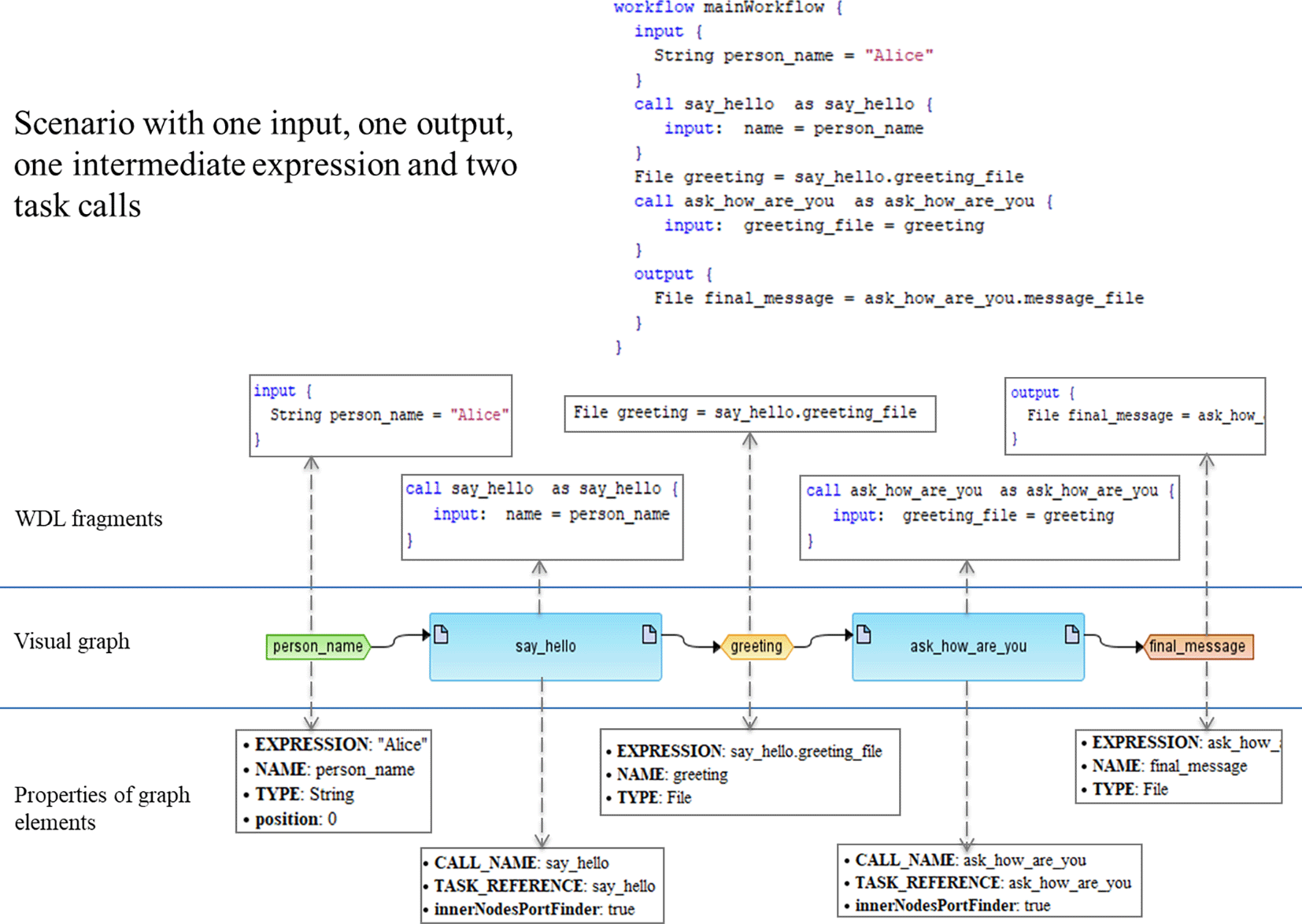
BioUML workflow graphical notation. In BioUML workflows are represented visually as graphs consisting of edges and nodes. Diagram elements and their notation is depicted in Table 2. Each diagram element corresponds to a part of the workflow. The diagram can be automatically generated on the basis of the WDL script. Vice versa, WDL or Nextflow may be generated from the diagram.
Graph layout. When we generate a visual diagram from text we also want to layout it automatically for user convenience and readability. BioUML provides a number of graph layout algorithms including hierarchic layouter suited for data flow representation, however it does not account for compartments (nodes inside nodes) in graph, while workflows may include nested cycles or conditional blocks of several level depth. On the basis of BioUML hierarchic layouter we have implemented a special layout algorithm for workflows which is performed as follows:
1. The graph is divided into interconnected subgraphs each in its own compartment.
2. We start with all subgraphs which do not contain nested compartments themselves and layout them with the standard BioUML algorithm.
3. We consider all nodes in the layouted subgraphs fixed and change the size of the compartment containing them to wrap all inner nodes. We substitute the original compartments by enlarged nodes.
4. If a layouted subgraph has edges connecting its inner nodes to other subgraphs we redirect all those edges from inner nodes to compartment enclosing subgraphs. Thus we obtain more high-level subgraphs without nested compartments.
5. Steps 2-4 are performed until all subgraphs are layouted.
6. We redirect all edges back to their initial inputs and outputs.
7. We perform BioUML built-in algorithm to generate layout for all edges in graph.
WDL parser. To generate a diagram from a WDL script we parse it into an abstract syntax tree using a parser generated with JavaCC grammar (https://javacc.github.io/). Then on the base of the abstract syntax tree we generate a diagram.
WDL and Nextflow generators. To generate Nextflow and WDL scripts from the diagram we use Velocity (https://velocity.apache.org/) template engine executed in Java code. Velocity provides structure of generated script and fills in data using java methods.
Web application and web form are accessible via any Internet browser. Users also may build their own instance of application from source code by performing following steps:
1. Install Java 21 (JRE or JDK).
2. Download source code from https://github.com/genespace-ru/workflow-engine .
3. Build workflow-engine using maven:
mvn package -DskipTests
4. Launch workflow-engine web edition with next command:
mvn jetty:run -Djetty.http.port=9998 -Dmaven.javadoc.skip=true -DskipTests=true
5. Use your Internet browser to open http://localhost:9998/
Additional information on how to use web applications and install applications is available at https://workflow-engine.readthedocs.io/en/latest/.
To test WDL to Nextflow conversion we have created a set of tests. Each test includes a WDL script with particular workflow elements and structures. For each WDL we test
1. Visual representation of script (WDL -> Diagram).
2. Round conversion (WDL -> Diagram -> WDL) with comparing input and output.
3. Nextflow conversion (WDL -> Nextflow) and compare with expected result.
4. Nextflow execution (WDL -> Nextflow -> Execution).
Examples of WDL diagrams may be found at https://workflow.genespace.ru/ in repository folder WDL Examples.
Command line application allows a user to:
- convert WDL workflow into Nextflow format;
- generate image in svg or png format for visual representation of workflow as graph using BioUML graphical notation (see “Materials and Methods”).
Documentation at workflow-engine.readthedocs.io describes how a user can install and use this program. Example of usage is provided on Figure 2.

Web service provides the same possibilities as the command line application but does not require a user to install the program. Documentation at workflow-engine.readthedocs.io describes how a user can use this service.
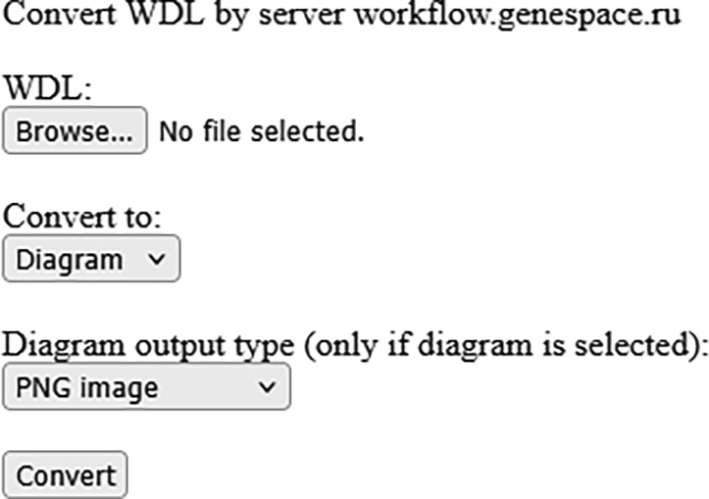
We also have developed a simple web form ( Figure 3) where user can upload files with WDL workflow and get corresponding workflow in Nextflow format and its picture. The form is available at wdl2nextflow.genespace.ru.
Web application ( Figure 4) provides possibilities to edit workflow both visually as a graph and as text. Users may import WDL scripts as well as input files and store them in a repository. WDL script from the repository can be opened as a visual diagram, properties of each diagram element (i.e. task, expression, cycle, etc.) can be edited by right clicking on the element, diagram may be layouted manually by moving elements. In the bottom part of the web interface a textual version of the workflow generated from the diagram is presented (both WDL and Nextflow versions). Users can change the generated WDL script in the lower panel and then automatically update the diagram according to changes. Finally the user can specify inputs for workflow from the repository and execute workflow from tab Settings.
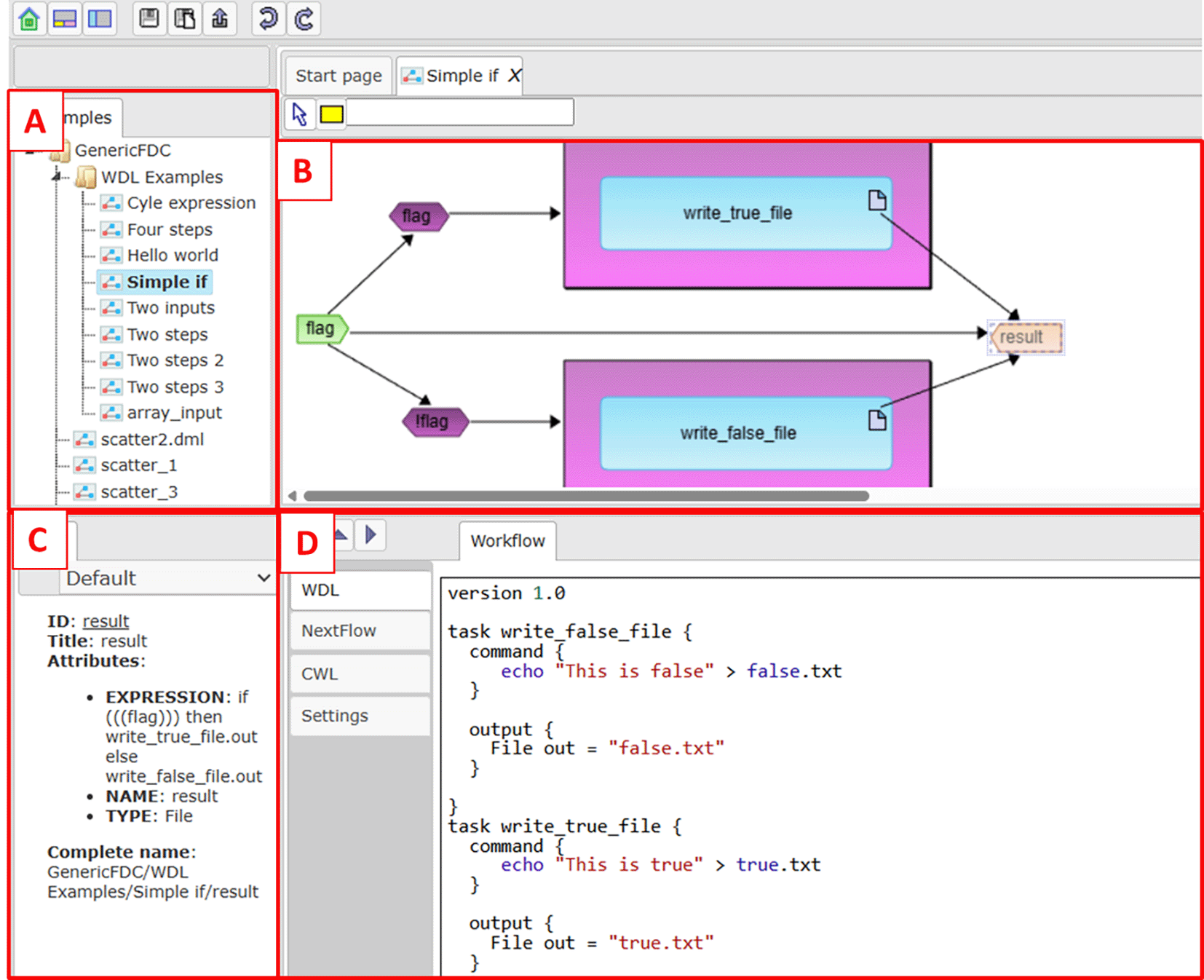
A - repository tree. B - Visual representation of the opened workflow. C - Properties of selected workflow element. D - Textual representation.
A full manual on how to work with the platform can be found at workflow-engine.readthedocs.io.
The developed plug-in integrates functionality described in previous section into BioUML platform ( Figure 5).
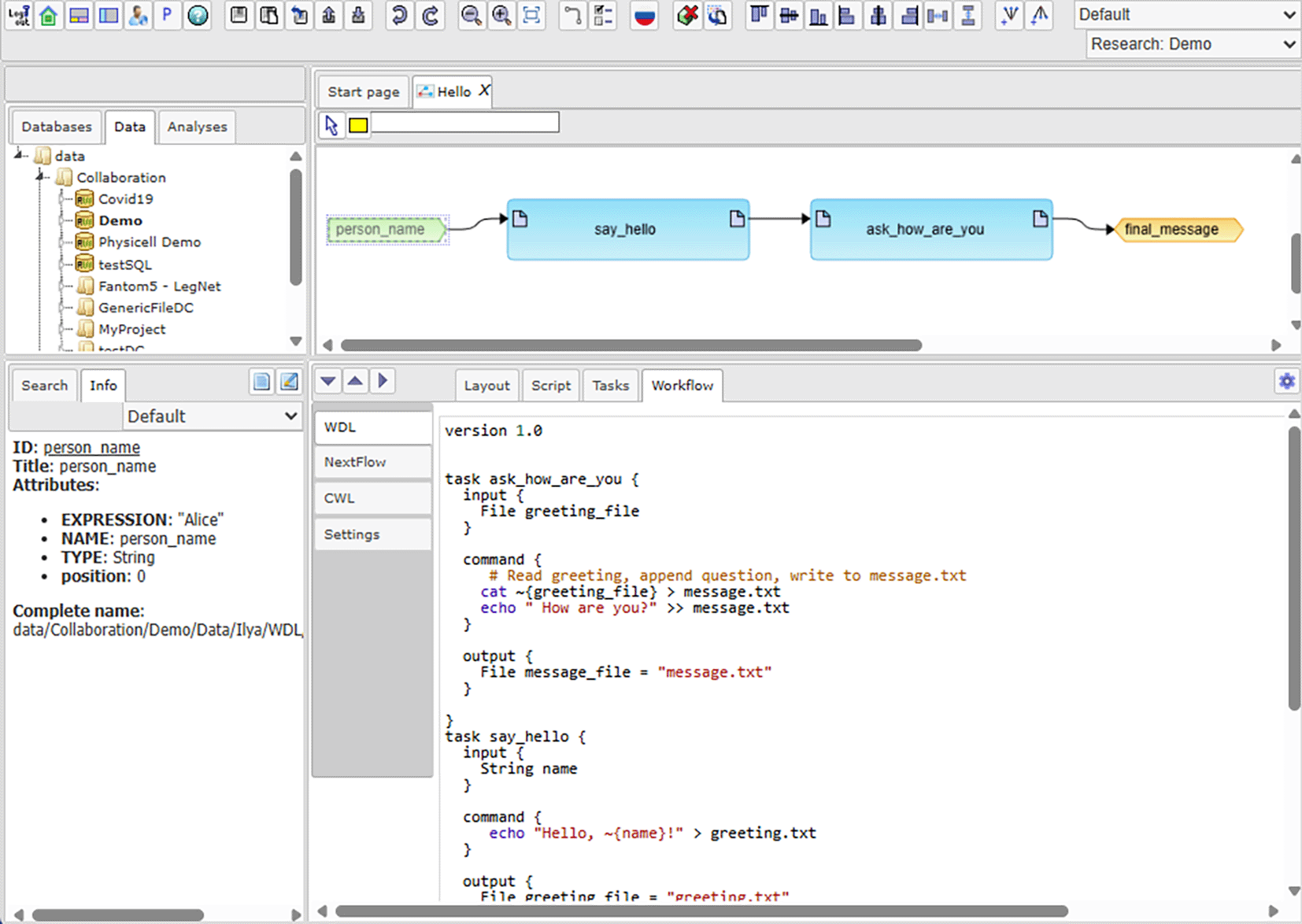
While both WDL and Nextflow describe pipelines of interconnected tools, their approaches are very different which makes conversion between them harder.
- WDL uses intuitive syntax and a static structure of workflow with explicit cycles (scatters), arrays, tasks, inputs and outputs.
- Nextflow on the other hand utilizes dataflow programming model approach, where data streams through channels that connect processes, cycles and arrays are implicit and hidden inside channels.
Our aim was to develop a set of universal rules for generating Nextflow scripts based on the data model created from WDL formalism (tasks, expressions, cycles and conditional blocks).
Description of tools connected into workflow (tasks in WDL and processes in Nextflow) are very similar in both languages and have the same structure with inputs, command, outputs and metadata, thus conversion is quite straightforward, with small changes to executing command itself necessary when WDL idioms are used (e.g. ~{sep=“ ” files} -> files.join()).
The main difficulty in conversion between WDL in Nextflow is dealing with arrays and cycles. WDL allows for very intuitive data manipulation while Nextflow tends to more idiomatic structures.
In Nextflow channels are executed asynchronously as elements are delivered via channel and order of outputs is not determined by order of inputs. To emulate WDL behavior of a determined output array we used a “fair true” directive for each process which forces output order to be the same as input order. Netflow also has arrays and in some cases WDL arrays are translated to Nextflow arrays while in others to Nextflow channels. In order to facilitate work with them we defined support Groovy functions such as get (array, i). length (array) that can work both with arrays and channels. In some cases we need to be sure that we work with channels (e.g. running call in cycle for elements of array) to that end we use the function to Channel (array) which returns a channel in any case (input can be both array or channel).
WDL defines a list of prebuilt functions such as “basename”, ”sub”, “read_int” and so on. To ease conversion we have created Groovy functions with the same name and functionality. All those functions are stored in supplemented biouml_function.nf script which is included into Nextflow converted from WDL. This allows us to minimally change expressions during conversion (see Table 3).
Range and length in nextflow are predefined Groovy functions to calculate length of array or channel and create channel with range. WDL - Workflow Description Language.
A full table of conversion rules and defined Groovy functions is presented at workflow-engine.readthedocs.io.
We have tested our approach on a limited number of tests for conversion of WDL scripts into Nextlow ones. So it is possible that there are cases where incorrect Nextflow will be generated. We will continue our work to create a more powerful test suite. Also some WDL workflows can be translated in a more concise way using Nextflow’s more complex structures.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.






Comments on this article Comments (0)