Keywords
Eucalyptus snout beetle, Illumina sequencing, Oxford nanopore sequencing, genome assembly, transcriptome, proteome prediction
This article is included in the Nanopore Analysis gateway.
This article is included in the Genomics and Genetics gateway.
Eucalyptus snout beetle, Illumina sequencing, Oxford nanopore sequencing, genome assembly, transcriptome, proteome prediction
Gonipterus species, or Eucalyptus snout beetles, are coleopterans that damage Eucalyptus trees in plantations globally (Tooke 1955). International trade has resulted in the introduction of Gonipterus species from their native ranges into other Eucalyptus-growing countries, impacting Eucalyptus plantation forests globally (Wingfield et al. 2008; Hurley et al. 2016; Schröder et al. 2020). Eucalyptus defoliation by Gonipterus scutellatus ranges between 5% and 80% of plantation trees, depending on the season and the management techniques used (Loch and Matsuki 2010). Reliable and sustainable management strategies are desperately needed to manage this forestry pest.
With the establishment of next-generation sequencing, the amount of genomic data available for model and non-model organisms, including Coleoptera, has increased exponentially. To date, there are 331 and 840 coleopteran genomes available on InsectBase (https://www.insect-genome.com/genome) and National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/), respectively, with these coleopterans most commonly classified as pests (Wang et al. 2025). While InsectBase and NCBI contain 33 and 106 curculionid genomes, respectively, this study represents the first genome of a curculionid from the genus Gonipterus (Wang et al. 2025). Coleopteran genomes exhibit substantial size variation, ranging from just 18.22 Mb in the scarabid Coptodactyla brooksi, to 2714.43 Mb in the lycid Platerodrilus igneus (Wang et al. 2025).
The availability of coleopteran genomes has facilitated the exploration of a broad range of research questions. Specifically, Curculionidae genomes have been used to investigate topics such as host adaptation, characterisation of symbiotic relationships, identification of genes involved in environmental tolerance, characterisation of range expansion, microsatellite mining, identification of chemoreceptor genes, and to determine population structure (Apriyanto and Tambunan 2021; Navarro-Escalante et al. 2021; Keeling et al. 2022; Mohd Rodzik et al. 2023; Wang et al. 2023; Biswas et al. 2024; Chen et al. 2024). From an evolutionary perspective, research has also focused on protein evolution, such as that of luciferin, which is involved in bioluminescence in fireflies (Zhang et al. 2020). In addition, these coleopteran genomes also allow comparative genomic studies to inform the development of gene-based pest control methods (Chu et al. 2018).
Detoxification is a crucial adaptive mechanism for insect pests, as it enables them to neutralise harmful substances, such as plant secondary metabolites and insecticides, enhancing their survival, prevalence, and ability to spread. In insects, cytochrome P450 monooxygenases (P450s) play a key role in the metabolic detoxification of xenobiotics, such as plant secondary metabolites and insecticides (Feyereisen 2012; Cui et al. 2016; Lu et al. 2021). These enzymes reduce the biological activity of toxic compounds by adding an oxygen atom, thereby neutralising their effects. The range of xenobiotics detoxified by insect P450s can vary from narrow to broad, while distantly related P450 enzymes may degrade the same compounds with differing efficiencies (Cui et al. 2016). Increased gene copy number or enhanced expression of P450 genes is often linked to greater resistance to insecticides (Feyereisen 2012). As a result, numerous studies have focused on identifying the specific P450 genes and gene superfamilies involved in detoxification, as well as their expression in response to insecticide exposure (Zhu et al. 2013; Evans et al. 2018; Zhang et al. 2023).
This article presents the first genome of Gonipterus sp. n. 2 (Coleoptera, Curculionidae), an important resource for better understanding the biology of this Eucalyptus pest and enabling further research into alternative pest control mechanisms.
A total of 57.1 Gb of short-read DNA sequencing data (151 bp paired-end reads) and 23.4 Gb RNA sequencing data were generated with the Illumina HiSeq platform. Long read PromethION sequencing yielded 76.41 Gb data with read N50 of around 22 kb. Genome profiling using short reads with GenomeScope 2.0 resulted in an estimated genome size of 1.8 Gb. The primary NECAT assembly had 2,589 contigs, an N50 of 2.6 Mb and an assembled genome size of 1.76 Mb. After polishing and purging of haplotypes, the final haploid genome assembly had 1,023 contigs, an N50 of 2.78 Mb and a haplotype genome size of 1.54 Gb ( Table 1). BUSCO analysis of the final assembly using the “insecta_odb10” dataset resulted in a completeness score of 98.3% (Manni et al. 2021).
RepeatModeler identified 4,917 repeat families in the genome, comprising 73.46% of the whole genome when masked with RepeatMasker. The Braker3 pipeline predicted 42,343 protein-coding genes with BUSCO score of 99%, higher than the scores obtained from BUSCO run on the assembly, indicating that the annotation has sufficiently covered the organism’s gene space. Protein clustering of Gonipterus sp. n. 2 predicted proteome with 13 other insect species (11 Coleoptera, one Lepidoptera and one Neuroptera ( Table 2)) using OrthoFinder resulted in 22,245 orthogroups. These species were selected based on the criterion that transcriptomic data were used as gene evidence during genome annotation, suggesting that their proteomes are of high quality. Of the identified orthogroups, 3,770 (16.95%) were shared by Gonipterus sp. n. 2, selected coleopteran species, and the two non-coleopteran species used as the outgroup ( Figure 1). A total of 1,398 (6.28%) orthogroups were unique to Gonipterus sp. n. 2.
| Insect species | Classification |
|---|---|
| Anoplophora glabripennis | Coleoptera, Cerambycidae |
| Altica viridicyanea | Coleoptera, Chrysomelidae |
| Bombyx moria | Lepidoptera, Bombycidae |
| Chrysoperla carneaa | Neuroptera, Chrysopidae |
| Dendroctonus ponderosae | Coleoptera, Curculionidae |
| Gonioctena quinquepunctata | Coleoptera, Chrysomelidae |
| Hypothenemus hampei | Coleoptera, Curculionidae |
| Ips typographus | Coleoptera, Curculionidae |
| Oryctes borbonicus | Coleoptera, Scarabaeidae |
| Protaetia brevitarsis | Coleoptera, Scarabaeidae |
| Rhynchophorus ferrugineus | Coleoptera, Curculionidae |
| Sitophilus oryzae | Coleoptera, Curculionidae |
| Tribolium castaneum | Coleoptera, Tenebrionidae |
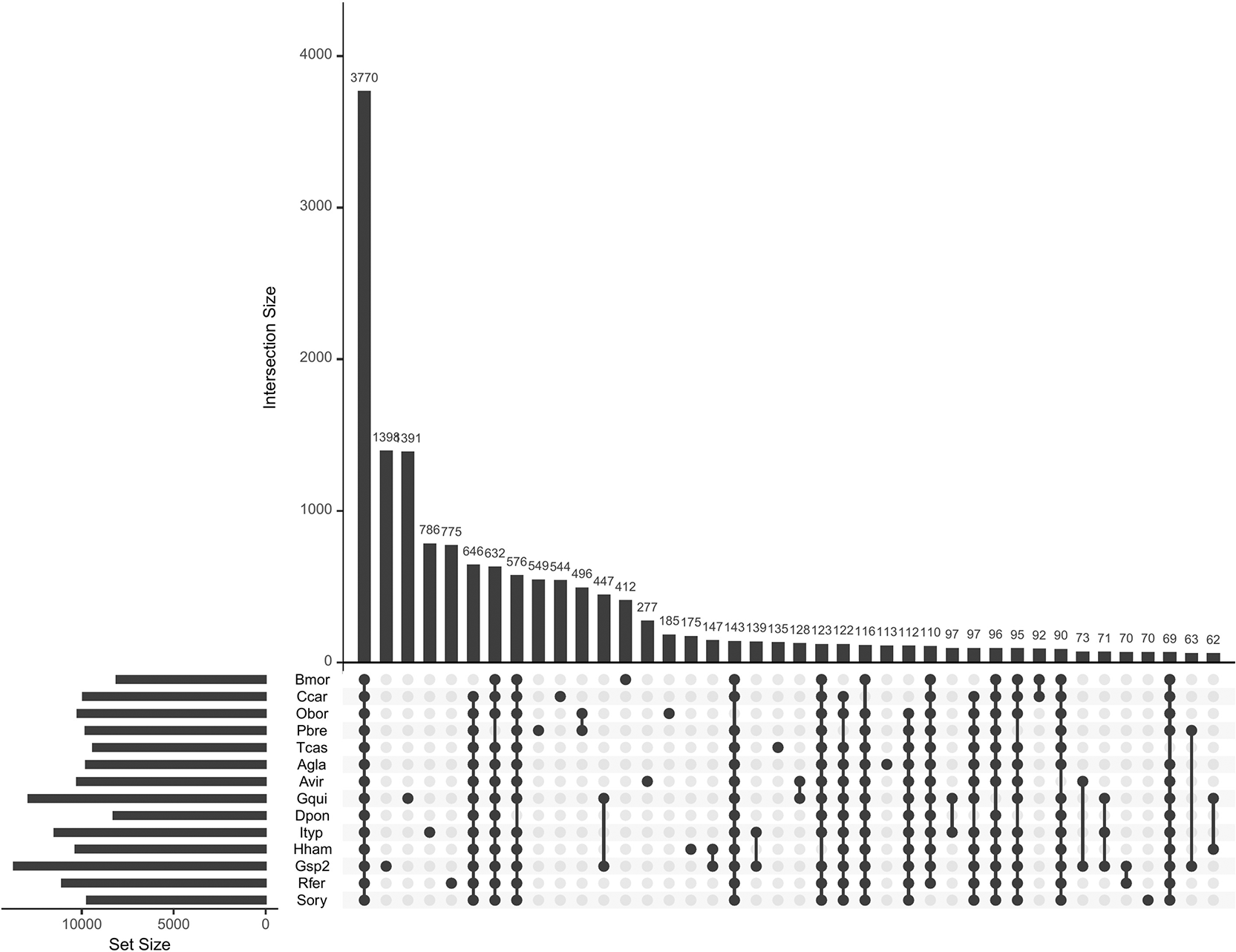
There were 3770 genes shared by all the species and the number of genes unique to an individual species was between 1398 and 70 genes by Gonipterus sp. n. 2 and S. oryzae, respectively. The set size for the species was between 5000 and 15000. The abbreviations for the species names are as follows Bmor was B. mori, Ccar was C. carnea, Obor was Oryctes borbonicus (Coleoptera, Scarabaeidae), Pbre was Protaetia brevitarsis (Coleoptera, Scarabaeidae), Tcas was T. castaneum, Agla was Anoplophora glabripennis (Coleoptera, Cerambycidae), Avir was Altica viridicyanea (Coleoptera, Chrysomelidae), Gqui was Gonioctena quinquepunctata (Coleoptera, Chrysomelidae), Dpon was Dendroctonus ponderosae (Coleoptera, Curculionidae), Ityp was Ips typographus (Coleoptera, Curculionidae), Hham was Hypothenemus hampei (Coleoptera, Curculionidae), Gsp2 was Gonipterus sp. n. 2 (Coleoptera, Curculionidae), Rfer was Rhynchophorus ferrugineus (Coleoptera, Curculionidae) and Sory was S. oryzae.
CAFÉ analysis of Gonipterus sp. n. 2 proteome together with those from 11 other coleopteran species, Bombyx mori (Lepidoptera, Bombycidae) and Chrysoperla carnea (Neuroptera, Chrysopidae) identified 437 gene families with significant expansion and 110 gene families with significant contraction ( Figure 2). One hundred and nineteen P450 genes were found in 22 gene families in Gonipterus sp. n. 2, while the remaining 13 species had between 45 and 105 cytochrome P450 monooxygenase genes ( Figure 2). Four cytochrome P450 monooxygenase gene families in Gonipterus sp. n. 2 experienced significant expansion and contained 79 (66.39%) of the identified cytochrome P450 monooxygenase genes ( Figure 3A– 3D). None of the cytochrome P450 monooxygenase gene families in Gonipterus sp. n. 2 experienced significant contraction.
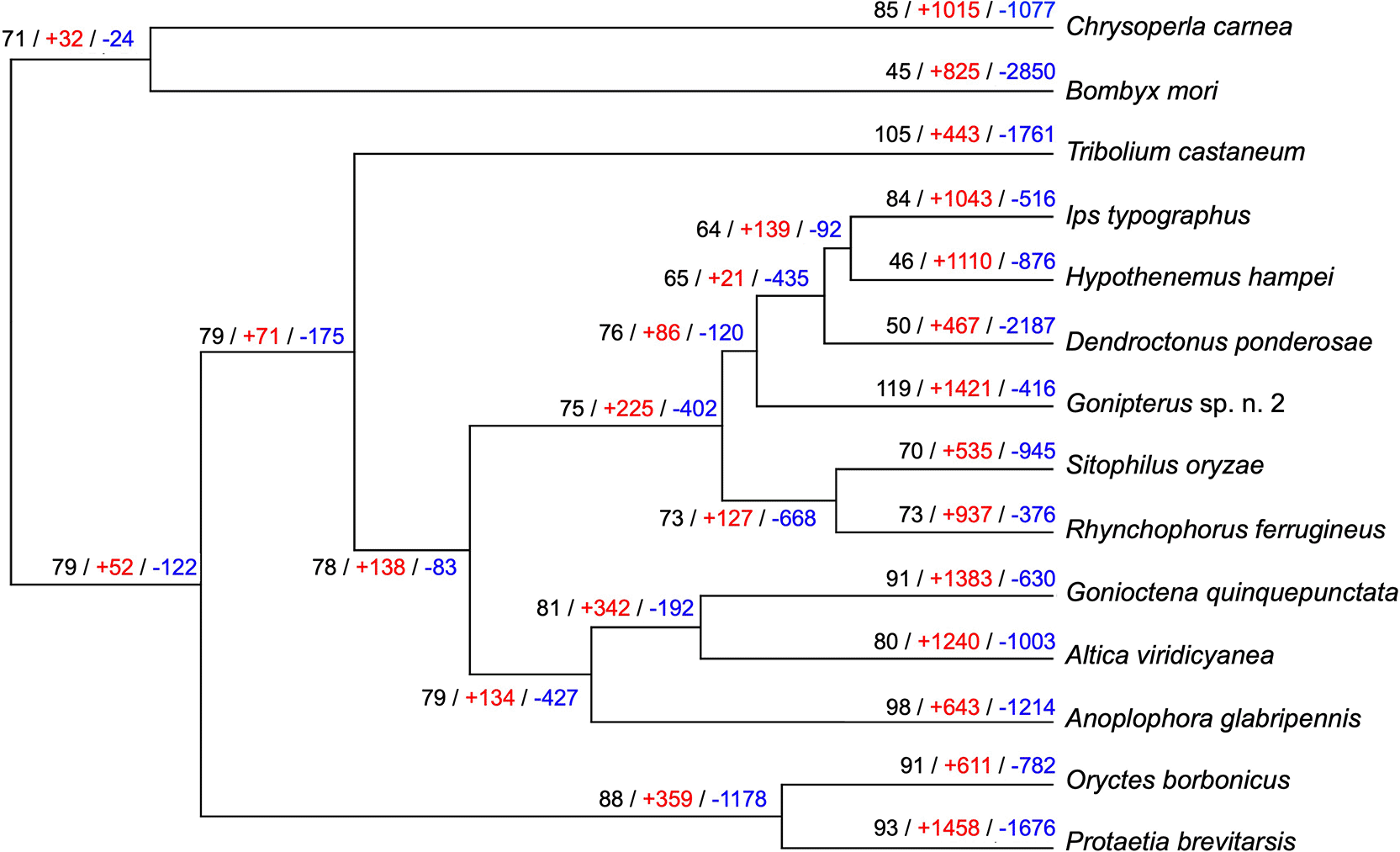
The largest number of cytochrome P450 monooxygenase genes, indicated in black, was identified in Gonipterus sp. n. 2 (n = 119). The number of cytochrome P450 monooxygenase genes identified in selected coleopteran species ranged from 46 to 105 genes. Chrysoperla carnea and B. mori had 85 and 45 cytochrome P450 monooxygenase genes, respectively. The number of gene families with significant expansion and contraction is indicated in red and blue. Gonipterus sp. n. 2 has the largest significant expansion and smallest significant contraction of gene families of the selected coleopteran species and the outgroups.
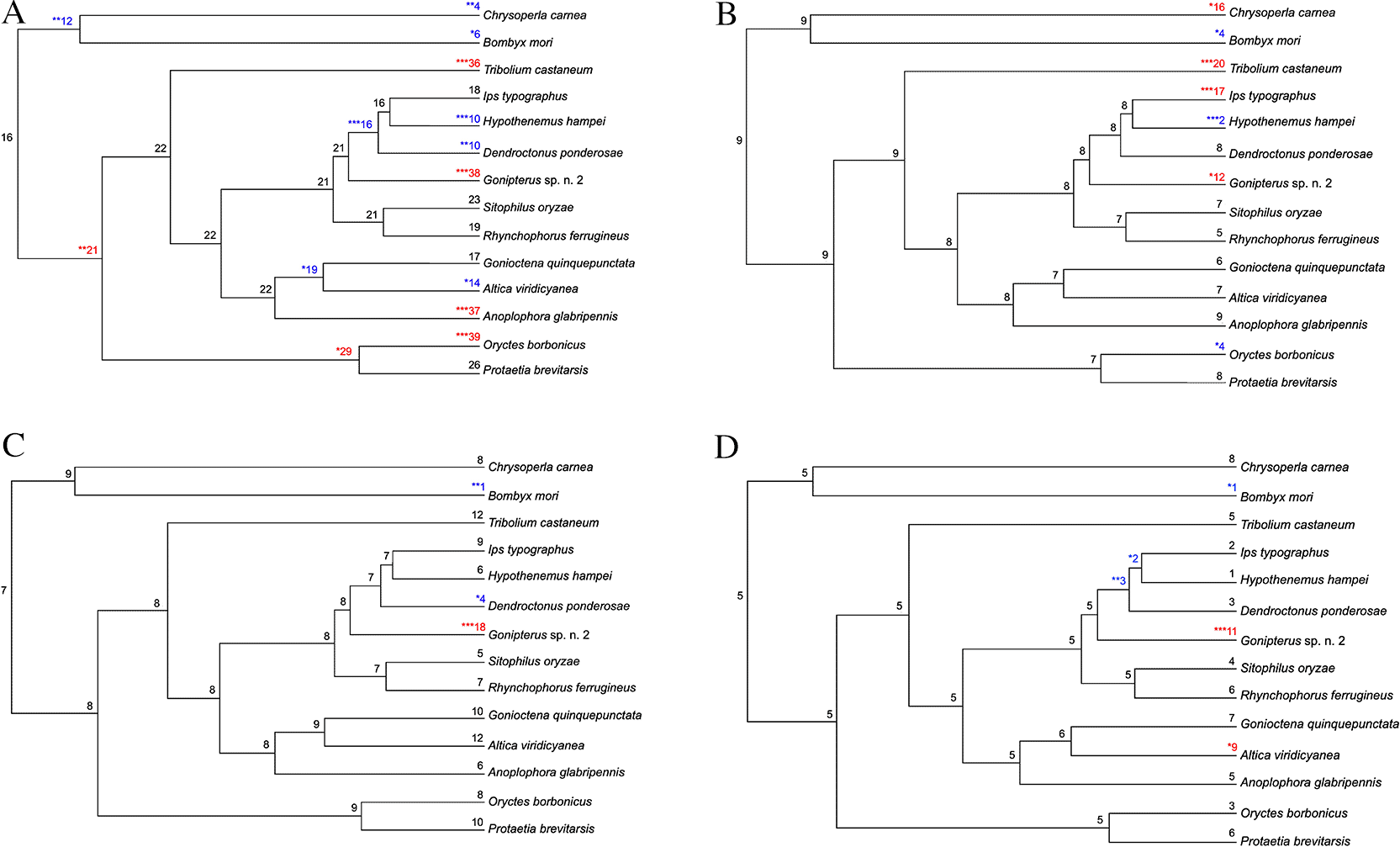
The cytochrome P450 monooxygenase gene family protein domain was identified in Orthogroups by the Blast2GO PRO plug-in on the CLC Genomics Workbench. The numbers in red and blue at the nodes indicate expansion and contraction of the orthogroups. The numbers in black at the nodes indicate no expansion or contraction occurred. The statistical significance of the expansion or contraction at the individual nodes is indicated with asterisks; *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Gonipterus sp. n. 2 had the highest number of cytochrome P450 monooxygenase genes (119 genes) amongst the species assessed. The expansion of cytochrome P450 monooxygenase genes may enable Gonipterus sp. n. 2 to detoxify the secondary metabolites in Eucalyptus leaves. Eucalyptus species have a high diversity of secondary metabolites with different biological activities, including insecticidal, antimicrobial, and antifeedant (Brezáni and Karel 2013; Danna et al. 2024). Insect-repellent and insecticidal activities of the Eucalyptus secondary metabolites were effective against different coleopterans, including Tribolium castaneum (Coleoptera, Tenebrionidae) and Sitophilus oryzae (Coleoptera, Curculionidae) (Danna et al. 2024). However, Gonipterus sp. n. 2 was attracted to the volatiles of damaged leaves from Eucalyptus host species (Bouwer et al. 2014). Increased diversity, copy number and expression of cytochrome P450 monooxygenase genes in insects is associated with increased xenobiotic resistance (Feyereisen 2012).
The Gonipterus sp. n. 2 genome may enable the development of alternative control methods, such as genetic pest control, to supplement currently applied pest control measures. Other coleopteran genomes have also been utilized to develop alternative pest control strategies. Specifically, these genomes have been leveraged to identify potential target genes for RNAi-based pest control and to facilitate CRISPR/Cas9 genome editing (Segers et al. 2023; Johny et al. 2024; Zhang et al. 2024). Such research in Coleoptera has primarily focused on model organisms, such as Tribolium castaneum, highly invasive species like Harmonia axyridis and insect pests of economic importance, such as Leptinotarsa decemlineata (Gui et al. 2020; Wu et al. 2022; Markley et al. 2024). Knowledge gained from the Gonipterus sp. n. 2 genome thus not only provides opportunities to direct future research to understand the beetle’s biology, but potentially also to improve and develop new pest control methods.
For long-read sequencing, Gonipterus sp. n. 2 adults were collected from Eucalyptus plantations near Greytown (KwaZulu-Natal, South Africa) (coordinates: 29.218415°S and 30.679624°E) during September 2021. The following research was carried out with approval from the University of Pretoria, Natural and Agricultural Sciences Ethics Committee (Reference number: NAS173/2020). The abdominal tissue only from a single adult was used for DNA extraction following the Monarch High Molecular Weight (HMW) DNA Extraction Kit (catalogue number T3060L; New England Biolabs, Ipswich, MA, United States of America) protocol for tissue, with slight modifications to the manufacturer’s instructions. The following modifications were made in Part 1: Tissue lysis. At step 5, the lysate mixture was incubated at 56 °C for 15 minutes with 1,150 rpm agitation, and, subsequently, 30 minutes without agitation to increase the DNA yield. During the 30 minutes incubation step without agitation, the lysate mixture was inverted every 5 minutes to ensure complete lysis. At step 9, the sample was centrifuged at 25 °C at 16,000 rcf for 15 minutes. At step 11, the DNA phase was aliquoted into a clean Eppendorf tube and centrifuged at 25 °C at 16,000 rcf for 15 minutes. After the second centrifugation step, the adipose was pipetted from the solution, and the DNA phase was transferred to a labelled Monarch 2 ml Tube. The DNA was separated from the solution and eluted following the HMW gDNA Binding and Elution procedure of the Monarch HMW DNA Extraction Kit protocol for tissue.
For short read sequencing, Gonipterus sp. n. 2 adults were collected from Eucalyptus plantations near Melmoth (KwaZulu-Natal, South Africa) (coordinates: 28.562856 °S, 31.191291 °E) during October 2020. The gut content was removed and DNA extraction was performed following the E.N.Z.A® Insect DNA Kit (catalogue number D0926-01; Omega Bio-tek, Norcross, GA, United States of America). An individual adult was frozen in liquid nitrogen and ground to a fine powder. Proteinase K (25 μl) and CTL Buffer (350 μl) (catalogue number D0926-01; Omega Bio-tek, Norcross, GA, United States of America) were mixed with the ground samples, and the mixture was incubated overnight at 37 °C. The DNA was isolated, and RNA was degraded following the E.N.Z.A® Insect DNA Kit protocol.
The Gonipterus sp. n. 2 genome was sequenced with the Illumina and Oxford Nanopore sequencing platforms. For Illumina sequencing, a paired-end library (350 bp median insert size) was prepared using TruSeq PCR-free protocol and sequenced on the HiSeq platform at Macrogen (Seoul, Korea) to obtain 151 bp paired-end reads. Read quality from the Illumina data was assessed with FastQC v0.11.9 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and low-quality bases and remaining Illumina adaptors were removed with Trimmomatic v0.38 (Bolger et al. 2014). For Nanopore sequencing, the library was constructed using the ligation sequencing kit (SQK-LSK110) and sequenced on the FLO-PRO002 flow cell (PromethION) at Centre for Genome Innovation at the University of Connecticut. Basecalling was conducted using Guppy v5.1.12.
For RNA sequencing, Gonipterus sp. n. 2 adults were collected from Eucalyptus species near Greytown (KwaZulu-Natal, South Africa) (coordinates: 29.1934795° S and 30.6082423° E) during January 2023. RNA extraction was performed using the gut tissue of the Gonipterus sp. n. 2 adults with the InviTrap® Spin Plant RNA Mini Kit (catalogue number 1064100400; Invitek Diagnostics, Germany) following the manufacturer’s instructions. Briefly, 10 samples, each containing the gut tissue from Gonipterus sp. n. 2 adults, were frozen in liquid nitrogen and ground into a fine powder. Cells were lysed by adding 900 μl of Lysis Solution RP (catalogue number 1064100400; Invitek Diagnostics, Germany). The mixture was incubated for 30 minutes at 55 °C and vortexed every five minutes. The DNA was removed by centrifuging the mixture at 16,160 rcf for one minute and filtering the resulting supernatant with a Prefilter. The RNA was bound to an RNA Spin Filter and washed following the manufacturer’s instructions. To elute the RNA, 30 μl of the Elution Buffer R (catalogue number 1064100400; Invitek Diagnostics, Germany) was added to the RNA Spin Filter, incubated for two minutes at 25 °C, and then centrifuged for one minute at 11,000 rcf. The eluted RNA was immediately stored at -80 °C. A paired-end RNA library was constructed using the TruSeq Stranded mRNA Library Prep Kit, and the library was sequenced with Illumina HiSeq sequencing to obtain 151 paired-end reads at Macrogen Europe (The Netherlands). Adaptors and low-quality RNA sequences of reads were removed with Trimmomatic v0.38.
The genome size and heterozygosity of Gonipterus sp. n. 2 were estimated from the trimmed Illumina data with JELLYFISH v.1.1.12 (Marçais and Kingsford 2011) and GenomeScope 2.0 (Ranallo-Benavidez et al. 2020), with a k-mer value of 21. The NECAT pipeline (Chen et al. 2021) was used to assemble the uncorrected reads from the Nanopore data using the estimated genome size obtained from GenomeScope and a minimum read length of 3 kb. The raw Nanopore data were mapped to NECAT assembly with minimap 2.0 (Li 2018), and the mapping file was used to polish the assembled genome with racon v1.3.1 (Vaser et al. 2017) for three iterations. The trimmed Illumina data were mapped to the racon-polished genome with BWA v0.7.17 (Vasimuddin et al. 2019) and used to further polish the genome with Pilon v1.23 (Walker et al. 2014) for three iterations. A final round of polishing was conducted with racon using trimmed Illumina data mapped to the Pilon-polished assembly. Haplotypes in the primary polished assembly were removed with Purge Haplotigs (Roach et al. 2018). The completeness of the polished genome was assessed with the Benchmarking Universal Single-Copy Orthologs (BUSCO) v.4.0.5 utilising the “insecta_odb10” dataset (Manni et al. 2021).
Structural annotation was carried out using the Braker3 pipeline (Stanke et al. 2006; Stanke et al. 2008; Buchfink et al. 2015; Hoff et al. 2019; Brůna et al. 2021), using Augustus as the gene predictor. RepeatModeler v2.0.2 (http://www.repeatmasker.org/RepeatModeler/) was used to construct a de novo repeat library, which was used to soft-mask the genome with RepeatMasker v4.1.2 (http://www.repeatmasker.org/RepeatModeler/). RNA sequencing data from Gonipterus sp. n. 2 reproductive tissue (NCBI accession number: SAMN19700001), alimentary tissue (NCBI accession number: SAMN19700000) (Souza et al. 2022), and the trimmed RNA sequencing data from the gut of Gonipterus sp. n. 2 (PRJNA1189815) were aligned to the masked Gonipterus sp. n. 2 draft genome with HISAT2 v2.2.1 (Kim et al. 2019). Proteomes of the selected coleopteran species ( Table 2) were mapped to the Gonipterus sp. n. 2 genome with GenomeThreader v1.7.4 (Gremme et al. 2005). The aligned transcriptomes and proteomes were used as evidences to train the ab initio prediction tool Augustus for the Braker3 pipeline. The Blast2GO PRO plug-in v1.20.14 on the CLC Genomics Workbench v20.0.4 was used to assign functions to the predicted proteins and identify P450 genes using the protein domain (Pfam = 00067).
Orthologous gene families from the protein sequences of Gonipterus sp. n. 2, selected coleopteran species ( Table 2), Bombyx mori (Lepidoptera, Bombycidae) and Chrysoperla carnea (Neuroptera, Chrysopidae) were determined using OrthoFinder v2.5.5 (Emms and Kelly 2015), with the Diamond in sensitive mode (-S) and the inflation factor of 1.5. The coleopteran species and outgroups were selected from InsectBase (Accessed in June 2023) (Mei et al. 2022) based on the availability of genomes annotated with transcriptomic data, which enabled the prediction of a higher quality and more comprehensive protein set (Chen et al. 2017). To construct a species phylogeny, protein sequences of the single-copy orthologs were aligned with muscle v3.8.31 (Edgar 2022), and trees were inferred with IQ-TREE v2.1.2 (Kalyaanamoorthy et al. 2017). A species phylogeny was inferred from individual gene trees with ASTRAL v5.7.7 (Zhang et al. 2018). The obtained phylogeny was rooted in B. mori and C. carnea, and the branch length was optimised with RAxML v8.2.11 (Stamatakis 2014) using the concatenate alignment of all single-copy genes. The rooted phylogenetic tree was transformed into an ultrametric tree and used as input in CAFÉ. CAFÉ v5 (Mendes et al. 2020) was used to assess the expansion and contraction of P450 gene families.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)