Keywords
protein folding; hydrophobicity; dimerization; induced folding; funnel model
This article is included in the Cell & Molecular Biology gateway.
This article is included in the Bioinformatics gateway.
protein folding; hydrophobicity; dimerization; induced folding; funnel model
The history of protein structure prediction monitored by CASP (Critical Assessment of Protein Structure Prediction) in two-year cycles has revealed advances in the prediction of the 3D structure of proteins with a given amino acid sequence.1,2 This project has led to many milestones, such as the identification of so-called new folds with previously unknown secondary and super-secondary structures.2 Progress has also been seen in the development of force fields and numerous tool packages: Amber3 CHARMM4 ROBETTA5 or Gromacs6 and many others. Those mentioned are often used in drug design projects, where the structure of the protein target molecule must be known in order to predict the the expected potential interaction with the designed drug.7,8 However, a significant advancements have been introduced by the artificial intelligence-based method AlphaFold in its several versions,9,10 where the predicted structure is in full agreement with the experimentally determined structure. This progress is undoubtedly appreciated by pharmacology or pharmacy precisely in the field of drug design.11
However, the mechanism for obtaining the functional spatial structure of a protein, on which its biological activity fully depends, still remains a mystery. The question of why the protein acquired a particular structure remains unanswered. To this end, the force fields used to describe interactions in the protein body are constantly being modified, introducing modern techniques to express the relevant types of interactions such as the particle-mesh Ewald electrostatics algorithms.12,13
The significant role of water, especially dynamics of water when coupled to proteins, their folding and activity, is also extensively considered. These issues discussed in form of review are presented in.14 The cited review addresses all aspects of the involvement of water in folding, stabilisation, reactivity of proteins and the relationship of water molecules and membranes including the process of penetration of single water molecules through the membrane. Attention is drawn to the vital importance of hydrogen bonding networks, also in the aspect of complex formation in particular. The analysis focuses on physical and chemical phenomena and the possibility of computer simulation of these phenomena. The basic form of taking into account the presence of water in the simulation of phenomena related to protein activity as well as the folding process is to represent the water environment with an appropriate (defined by the conditions of the simulated object) number of individual water molecules. The change in their ordering at the protein contact surface and their involvement in the processes in question is monitored.14
In the model proposed in this article, the aqueous environment is treated as an external force field directing and ordering the distribution of the relevant protein components including the relationship of polarity to hydrophobicity in particular.
The model used, referred to as the fuzzy-oil drop model, is a modification of the oil drop model introduced by Kauzmann.15 The modification involves replacing the discrete (two-layers) form of the description of the hydrophobicity distribution with a continuous function in a 3D Gaussian form spanning the protein body.16 Treating the hydrophobicity distribution as continuous in this way makes it possible to monitor and quantify local inconsistencies with the expected distribution preferred by the water environment (centralisation of non-polar residues with exposure of polar residues).
The postulated need for attention and concentration of research on the properties of water as such reported in14 is expressed by water property tests,17–19 including the possibility of water structuring,20–22 as well as the specificity of water structuring at the air contact surface.23 The interaction of water with a hydrophobic surface that does not exclude even the levitation phenomenon of water molecules in contact with a hydrophobic surface is an object of analysis.24 The effect of changing the characteristics of the solutions around the proteins was also found to be significant.25,26
The current work proposes to record the presence of the water environment on the folding process in the form of an external force field provided by water. Orientation of the protein folding process by this environment results in the hydrophobicity distribution in the protein body as represented by the 3D Gaussian function. The folding of a protein referred to as ultra-fast-folding was considered, demonstrating that its structure is precisely a perfect reproduction of the distribution of hydrophobicity in the protein body consistent with the orientation derived from the aqueous environment. The specific mechanism of homo-dimer formation is also discussed. A distribution of hydrophobicity as expected for an water environment and also a distribution guaranteeing biological function was identified in the SH3 domains. Using the example of this protein complexing the relevant polypeptides, the role of this domain as a provider of a local external force field is demonstrated.
An analysis of the effect of so-called induced folding is also given, where the required presence of a protein that ensures that the haemoglobin Alpha chain acquires a structure prepared for interaction with the Beta chain.
The object of analysis is a de novo designed protein dimer with the structure available in PDB ID-2GJH.27 The analysis of structural changes related to dynamics presented in28 is concerned with the structural changes and stability of this dimer under different external conditions.
Our analysis presents an assessment of the structures in question from the point of view of hydrophobicity distribution in monomers as well as in dimers, taking into account the environment, using a fuzzy oil drop model (FOD-M) to assess the structure.16
Table 1. presents a list of proteins and their complexes discussed herein.
| Protein | PDB ID | Source org. | Ref. |
|---|---|---|---|
| De novo designed | 2GJH | 27 | |
| Villin 1 | 2F4K | Gallus gallus | 29 |
| SH3 | 1S1N | Homo Sapiens | 30 |
| SH3 | 1TG0 | Saccharomyces cerevisiae | 31 |
| SH3 | 1WDX | Saccharomyces cerevisiae | 32 |
| SH3 + peptide | 1ZUK | Saccharomyces cerevisiae | 33 |
| SH3 | 1X2P | Homo Sapiens | 34 |
| SH3 + peptide | 2VKN | Saccharomyces cerevisiae | 35 |
| AHSP + α-chain of Hb | 1Y01 | Homo Sapiens | 36 |
| SH3 + peptide | 2LCS | Saccharomyces cerevisiae | 37 |
The phenomenon of the spontaneous process of formation of structures referred to as micelles relies on the variable interaction of parts of bi-polar molecules in an aqueous environment. The polar parts of the molecules locate on the surface providing an entropically favourable arrangement with the surrounding water. The hydrophobic parts locate in the centre of the system, creating a concentration of hydrophobicity – hydrophobic core - isolated from water contact.
By interpreting the structure of amino acids as bi-polar molecules with a varying ratio between the polar and non-polar parts, the process of protein folding can be compared to the process of micellization.
This trend was identified by the oil drop model introduced by Kauzmann.15 The current version of the oil drop model changes the discrete two-layer system – polar shell and hydrophobic core – into a continuous form. The level of hydrophobicity varies from zero at the surface to the highest concentration in the centre of the system. This type of distribution is expressed by a 3D Gaussian function spanning the body of the protein expressed by the function:
However the actual hydrophobicity distribution is the result of an inter-residual hydrophobic interaction expressed by a function introduced by M. Levitt38:
Both of these normalised distributions (the first expressions in eq. 1 and eq.2) can be compared using the Kullback-Leibler divergence entropy40:
where distribution P is the distribution under consideration – in the FOD-M model it is distribution O (eq. 2), distribution Q – is the reference distribution – in the FOD-M model, it is distribution T.
Distribution T is an idealised, perfect distribution consistent with an ideal micelle with a centrally located hydrophobic core and a polar surface. Hydrophobicity levels in the protein body decrease continuously from a maximum in the centre to zero at the surface.
However, the value of DKL (O|T) cannot be interpreted directly (entropy). Therefore, a second reference distribution R is introduced, where each Ri value assigned to successive effective atoms is equal and expressed as Ri = 1/N where N is the number of amino acids in the protein. This reference distribution expresses a constant, non-variable distribution in a protein lacking a hydrophobic core.
The relationship DKL(O|T) < DKL(O|R) indicates an approximation of the O distribution to the T distribution, which is interpreted as the presence of a hydrophobic core in the protein under consideration. To avoid describing one object with two parameters, the RD (Relative Distance) parameter was introduced, defined as:
An RD value <0.5 indicates the presence of a hydrophobic core.
Protein analysis confirms the validity of the adopted model. Indeed, groups of proteins with very low RD values (<0.3) have been identified. This group includes proteins: fast-folding. Ultra-fast-folding, down-hill and antifreeze type II proteins.41 The results of the simulation performed, taking into account the environment, indicate the need for a suitable module in the programmes to express the contribution of the environment to the procedures simulating the protein folding process.42 Other groups of proteins, e.g. membrane proteins, show RD values >0.5.
The water environment directing the folding process towards the generation of a micellar system is not the only one in which proteins show their activity. Membrane proteins, for stability in a hydrophobic membrane environment during folding, expose hydrophobic residues by concentrating hydrophilic residues in the centre (especially in the case of proteins acting as an ion channel).
In this situation, the expected hydrophobicity distribution takes the inverse form:
Index n denotes normalisation.
After analysis of numerous proteins, it became apparent that the distribution of hydrophobicity in the protein body mapped a distribution that was the sum of both functions:
The contribution of the aquatic environment is corrected by the presence of other components that reduce the polar character of the environment. The contribution of the modifying factor varies. This is expressed by the value of the K parameter, which for the proteins mentioned above (down-hill) is equal to K = 0.0 or very low K < 0.4. This means that the folding of a given protein takes place with the active participation of only the aqueous environment directing the folding process towards the generation of a micelle-like system.
The value of the K parameter is determined by an iterative procedure identifying the minimum value of DKL(O|M) (Fig. 1.C). The principle of the model described is illustrated in Fig.1.
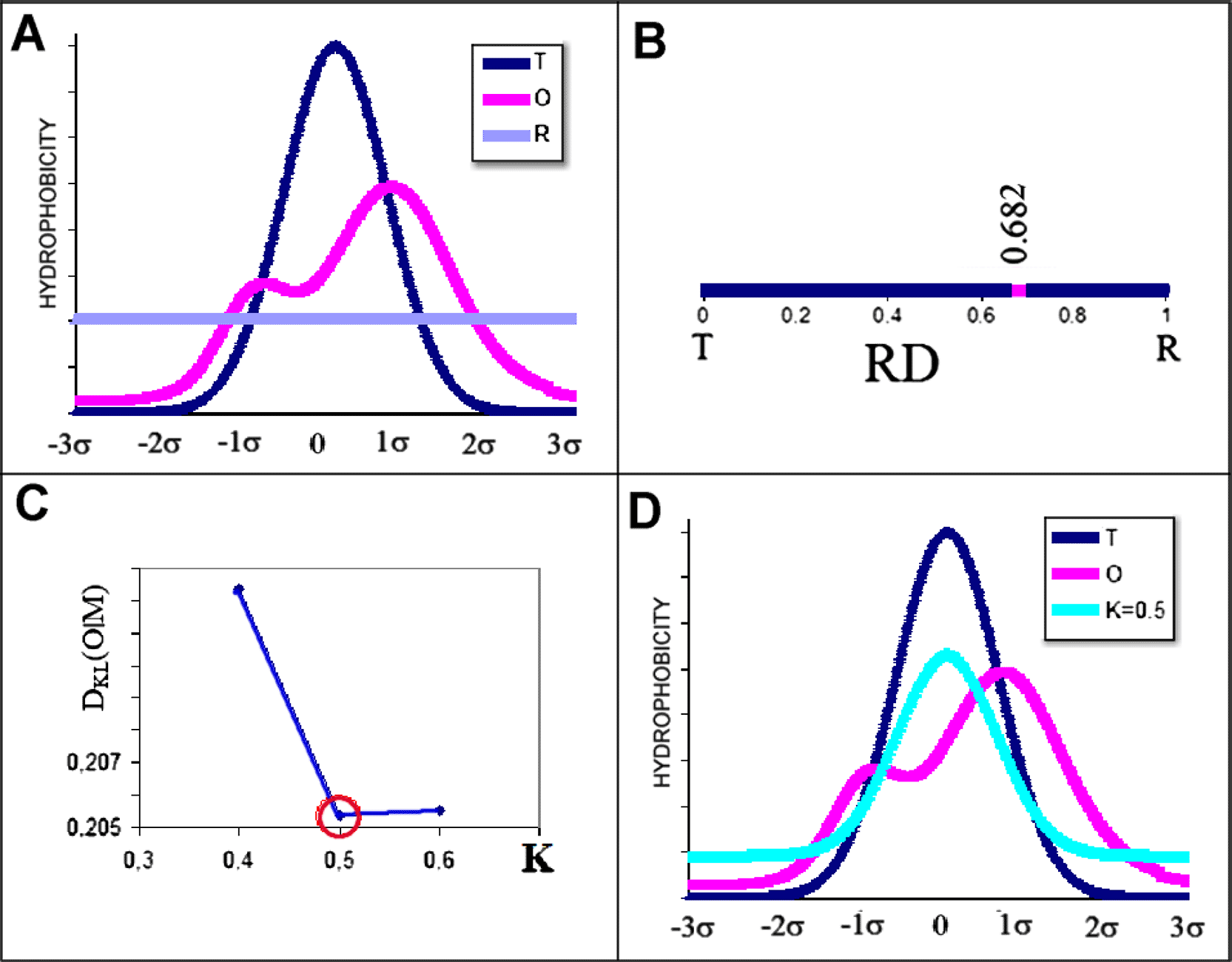
A – distribution T (blue), O (pink) and R (green).
B – the value RD = 0.682 determined for the system in A, indicating the absence of a hydrophobic core.
C – determination of the value of K by identifying the minimum value of DKL(O|M) for the set in A.
D – profiles T (blue), O (red) and M (cyan) for K = 0.5.
Interpretation of the FOD-M model parameters:
1. The RD value indicates the degree to which the hydrophobicity distribution in the protein body is aligned with the idealised micelle-like distribution (maximum hydrophobicity in the centre with zero hydrophobicity on the surface). Low values of RD < 0.5 indicate the presence of a hydrophobic core
2. The value of the K parameter indicates the contribution of other non-aquatic factors that make up the environment in which the folding takes place. The value K = 0.0 indicates that the folding process is directed solely by water.
An increase in RD > 0.5 expresses the local mismatch of hydrophobicity distribution O against distribution T. Elimination of residues showing significant Ti > Oi relationships (local hydrophobicity deficit) in a step-wise procedure may lead to RD < 0.5. This means identifying the part of the protein with a distribution locally consistent with a micelle-like distribution. Eliminated residues identify the position of the cavity prepared to interact with the ligand or substrate in the case of an enzyme. The relationship Ti < Oi means that the residue in question shows an increased level of hydrophobicity against the expected idealised one (local excess). If the residue is located on the surface, it means that an amino acid prepared to interact with an amino acid of another protein with a similar situation has been identified. The relationship Ti < Oi signals the exposure of hydrophobicity prepared to interact with another protein to form a complex.
The values of the RD and K parameters can be determined to an arbitrary structural unit: complex, single protein, domain. In these cases, a 3D Gaussian function is generated for each listed object. On the other hand, the contribution of an entity to the construction of a complex structure can also be determined: e.g. the contribution of a chain to a complex, the contribution of a domain to the structuring of a chain. The T, O and R profile fragment is then normalised for the selected fragment without the need to build a separate 3D Gaussian function.
It is also possible to determine the status of the residues involved in the construction of the complex by analysing the status of the interface. Residues remaining in contact can be eliminated from the T, O and R profiles of the complex. By determining the status of the interface thus isolated, it is possible to determine whether it has been formed on the basis of the construction of a common hydrophobic core. In this case, the status for the isolated interface is described by low RD values.
The program to calculate the RD and K parameters is available for free at https:\hphob.sano.science.
The experimental investigation (according to29) of subdomain of the chicken villin headpiece indicates that the substitution of two lysines by norleucine increased the folding rate 6-fold at 300 K and amounts to 0.7 micros. \.
The structure of this domain is available in the PDB resources – ID 2F4K.29 When analysed based on the hydrophobicity distribution (using the FOD-M model), it shows an alignment of the O distribution with the T distribution. This means that a centrally located hydrophobic core with a polar surface is present in the structure of this domain. A value of RD = 0.295 and K = 0.0 indicates an almost perfect reproduction of the hydrophobicity distribution that results from the folding process being directed by the water environment (Fig. 2).
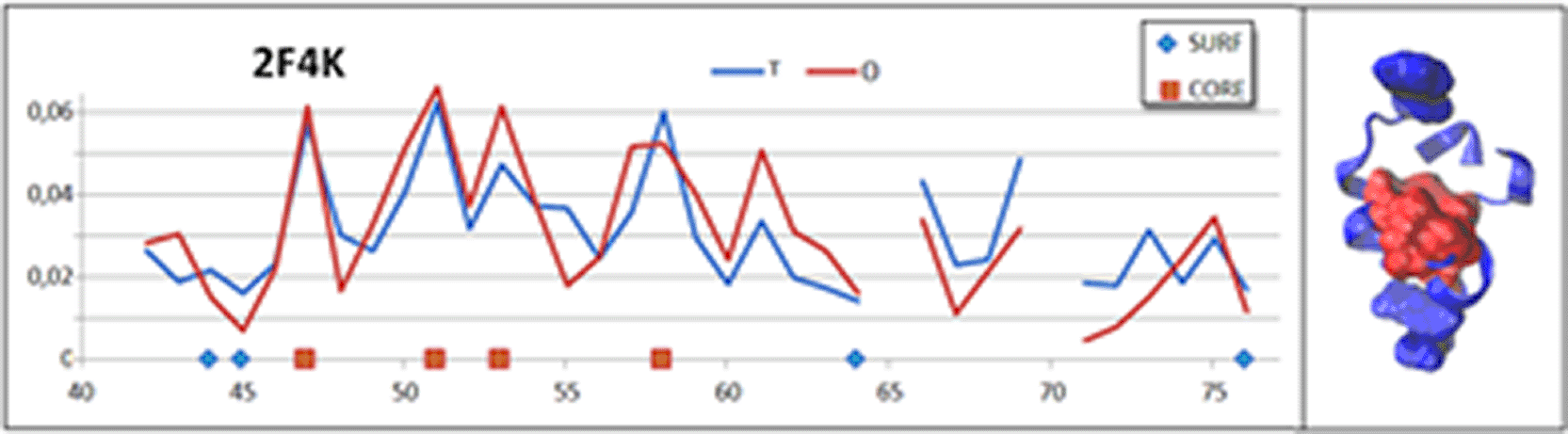
The M profile aligns with the T profile, as the determined value of the K parameter for this hydrophobicity distribution is K = 0.0. The 3D presentation distinguishes between the residues comprising the hydrophobic core (red spacefilling) and the residues representing local minima (blue spacefilling). The residues highlighted on the 3D presentation are also indicated on the horizontal axis of the profile set.
The determined residues responsible for reconstructing the local maxima forming the hydrophobic core are: 47F, 51F, 53 M, 58F (Fig. 2 – red squares). These residues were visualised on the 3D presentation (Fig. 2) as red squares on the horizontal axis of the hydrophobicity distributions and red space filling on the 3D presentation.
From the point of view of an interpretation based on the FOD-M model, the speed and ease of the folding process of this domain in an aqueous environment is not surprising.
Such results were also obtained for a number of other fast-folding, ultra-fast-folding, down-hill and antifreeze type II proteins.41
Complexation-dimerisation analysis, based on in silico methods, determines the conditions for the dimerisation process determined based on molecular dynamics simulations28 for both monomers and dimers of the Top7-CFr protein (PDB ID – 2GJH27).
Experimental studies in28 have shown the conditions leading to partial unfolding and refolding by operating the force field of the CHARM22 program for a temperature of 400 K – a temperature close to the experimentally observed process referred to as melting. In the monomer-containing box, The 6574 water molecules were located, while in the dimer-containing box – 12,497 water molecules. The presence of 0.1 M NaCl corresponding to 0.1 M of this salt. As a result, the protein concentration is expressed as a concentration of ∼8 mM.28
In the cited simulation, the presence of water is recorded by interaction with amino acid atoms located on the surface of the protein coming in contact with the atoms of water molecules. A description of monomer-folding phenomenon, starting from the extended form and corresponding to the experimentally available form28 (PDB ID - 2GJH), was obtained. Forms of intermediates leading to a structural form consistent with the experiment were achieved. In the analysis, some differences to the experimental observations were observed. It also concludes that “overall folding may depend on external conditions.” This observation is important for assessing the type of stabilisation of both monomers and dimers.
The results of the monomer and dimer structure analysis of the Top7-CFr protein in the FOD-M model assessment are given in Table. 2.
P-P position – status of residues included in the interface. No P-P – part of a structural unit with eliminated residues included in the interface.
| Structural units | P-P | No P-P | ||
|---|---|---|---|---|
| Individual units | RD | K | RD | RD |
| Chain A | 0.350 | 0.1 | 0.164 | 0.396 |
| Chain B | 0.369 | 0.1 | 0.426 | 0.387 |
| Dimer | 0.404 | 0.2 | 0.344 | 0.434 |
| Chains in complex | RD | K | ||
| Chain A | 0.364 | 0.1 | ||
| Chain B | 0.427 | 0.2 | ||
The RD and K parameter values given in Table. 2 reveal a very high ordering of hydrophobicity in the protein body consistent with the idealised distribution. This means obtaining a structure that is the result of the influence of the aqueous environment on the structuring process, consistent with the orientation of this type of external force field for both monomer and dimer structuring (Fig.3. and Fig. 4.). This high ordering applies both to chains treated as individual structural units (3D Gaussian functions generated for each unit individually) and to chains treated as components of a complex (assessment of the status of the T, O and M (for K = 0.2) profile fragments for chains, respectively) (Fig. 4.).
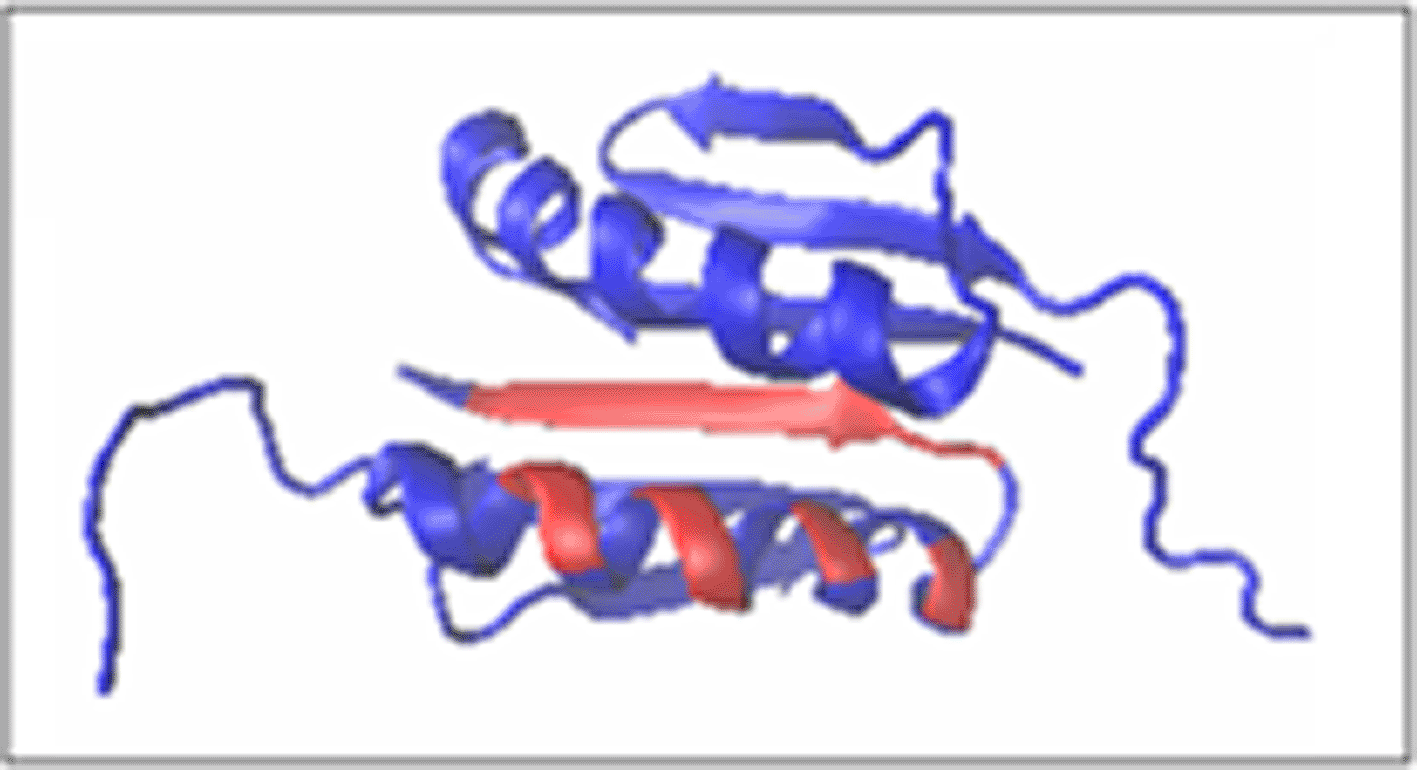
Chain B – the same residues interacting due to symmetry of the dimer.
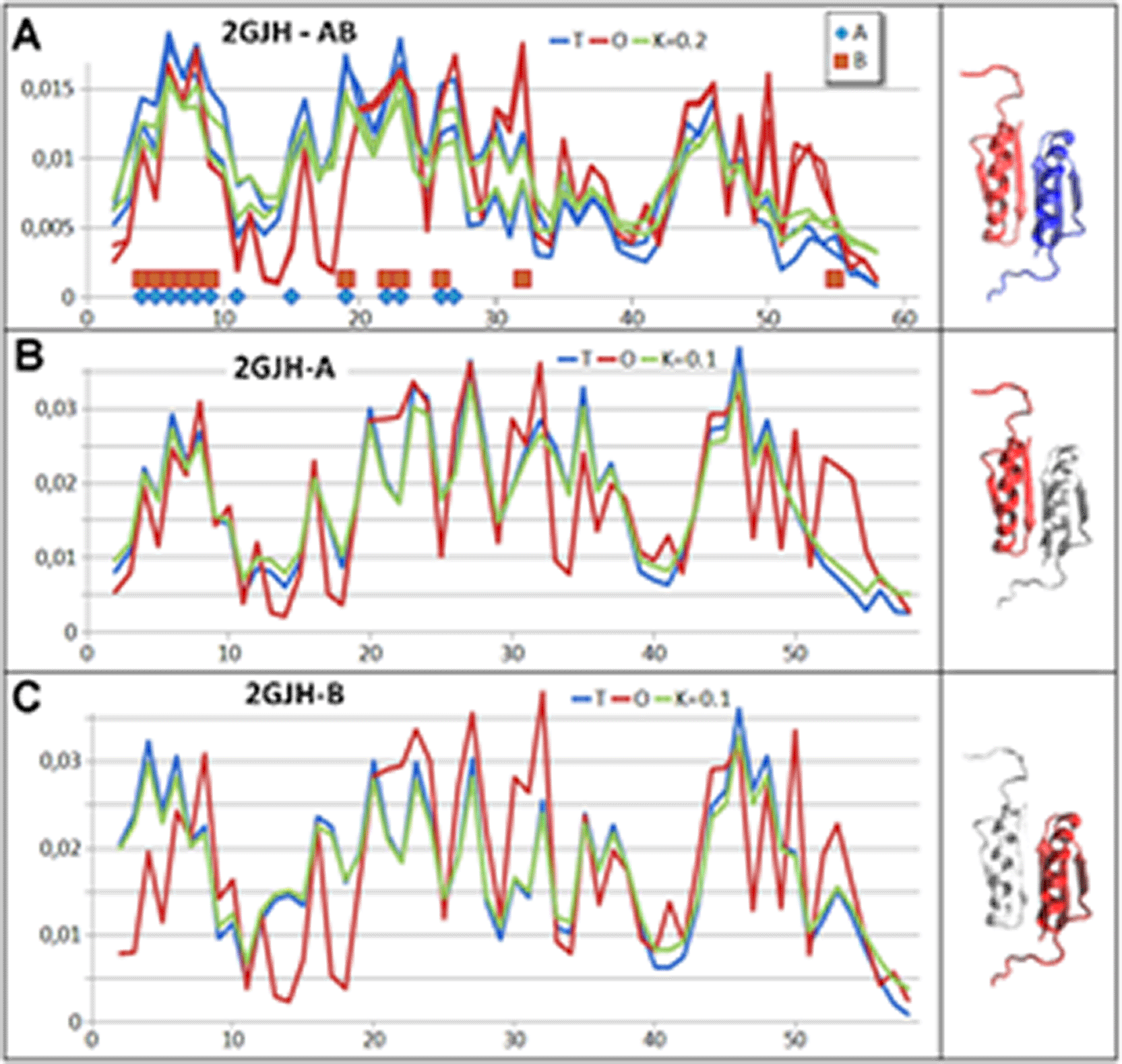
A – profiles T (blue), O (red) and M for K = 0.2 for the dimer. The horizontal axis distinguishes residues interacting with the second chain.
B – profiles T (blue), O (red) and M for K = 0.1 for the monomer – chain A. The horizontal axis indicates residues interacting with chain B.
C – profiles T (blue), O (red) and M for K = 0.1 for the monomer – chain B. Residues interacting with chain A are highlighted on the horizontal axis.
The assessment of the structural stability of the Top7-CFr monomers as well as the dimer is due to the clearly high ordering of the hydrophobicity distribution consistent with a micelle-like arrangement. The presence of a hydrophobic core is identified as a factor in the stabilisation of the protein’s tertiary structure (in addition to the presence of SS-bonds absent here). In the case of the protein in question, stabilisation is definitely based on the presence of a clearly defined hydrophobic core for both the monomers and the dimer.
The status of the monomers is much closer to micelle-like structuring in the FOD-M model classification, which is assumed to be due to the influence of the aqueous environment as a factor directing the folding process towards generating a hydrophobic core with a polar outer shell. P-P status – interface status – in the dimer, it represents the effect of a joint involvement in building dimer stabilisation – a very low RD value for the interface. This means that the interacting residues form a common core for the dimer.
Examples of other proteins with a structure associated with the presence of a micelle-like hydrophobicity distribution include the SH3 domain. Table. 3. presents a set of examples demonstrating a high degree of micelle-like ordering of hydrophobicity.
| DOMAIN | CHAIN | RD | K |
|---|---|---|---|
| 1TG0 | 0.350 | 0.1 | |
| 1WDX | A | 0.361 | 0.1 |
| B | 0.357 | 0.1 | |
| C | 0.347 | 0.1 | |
| D | 0.345 | 0.1 | |
| 1ZUK | A | 0.340 | 0.0 |
| B | 0.350 | 0.1 | |
| 1S1N | 0.311 | 0.0 | |
| 1X2P | 0.467 | 0.3 | |
| (1–60) | 0.423 | 0.2 |
This domain represents a conservative (from prokaryotes through viruses and eukaryotes) protein module. It plays a role in cellular signalling processes by controlling inter-protein interactions. This is of particular relevance to cytoskeletal structure or kinase activity.
In the light of the assessment of structuring based on the FOD-M model of the SH3 domain, the structure of this domain represents another example of a hydrophobicity distribution adapted to the conditions imposed by the aqueous environment leading to an ordering of hydrophobicity according to the micelle-like model. A centrally located hydrophobic core with a polar surface promotes the stabilisation of the structure achieved by this domain and ensures the functioning of this domain.
This term indicates the need for one of the monomers to be present for the folding of the other chain. One of the chains is a target for the folding and for the adjustment of the partner in the complex.
Signalling protein 2LCS37 – a protein that binds multiple targets – peptides that show similar strong affinity. In the example discussed here, the SH3 domain interacts with a 16-amino-acid peptide with serine/threonine-protein kinase STE20 from Saccharomyces cerevisiae.
From the point of view of the applications of the FOD-M model, this example forms the basis for the analysis of induced folding.
The SH3 domain is a target for the peptide, which in the interpretation of the FOD-M model means that the SH3 domain provides an external force field imposing the structuring of the peptide. The peptide in question, due to its small number of amino acids, is unable to produce its own tertiary structure and adapts relatively easily to the target, which in this case is the SH3 domain. Nevertheless, using the in silico procedure for protein and peptide folding, the introduction of an external force field in the form of the parameter K = 1.5 would arguably provide the structure represented in this case (Fig. 5.).
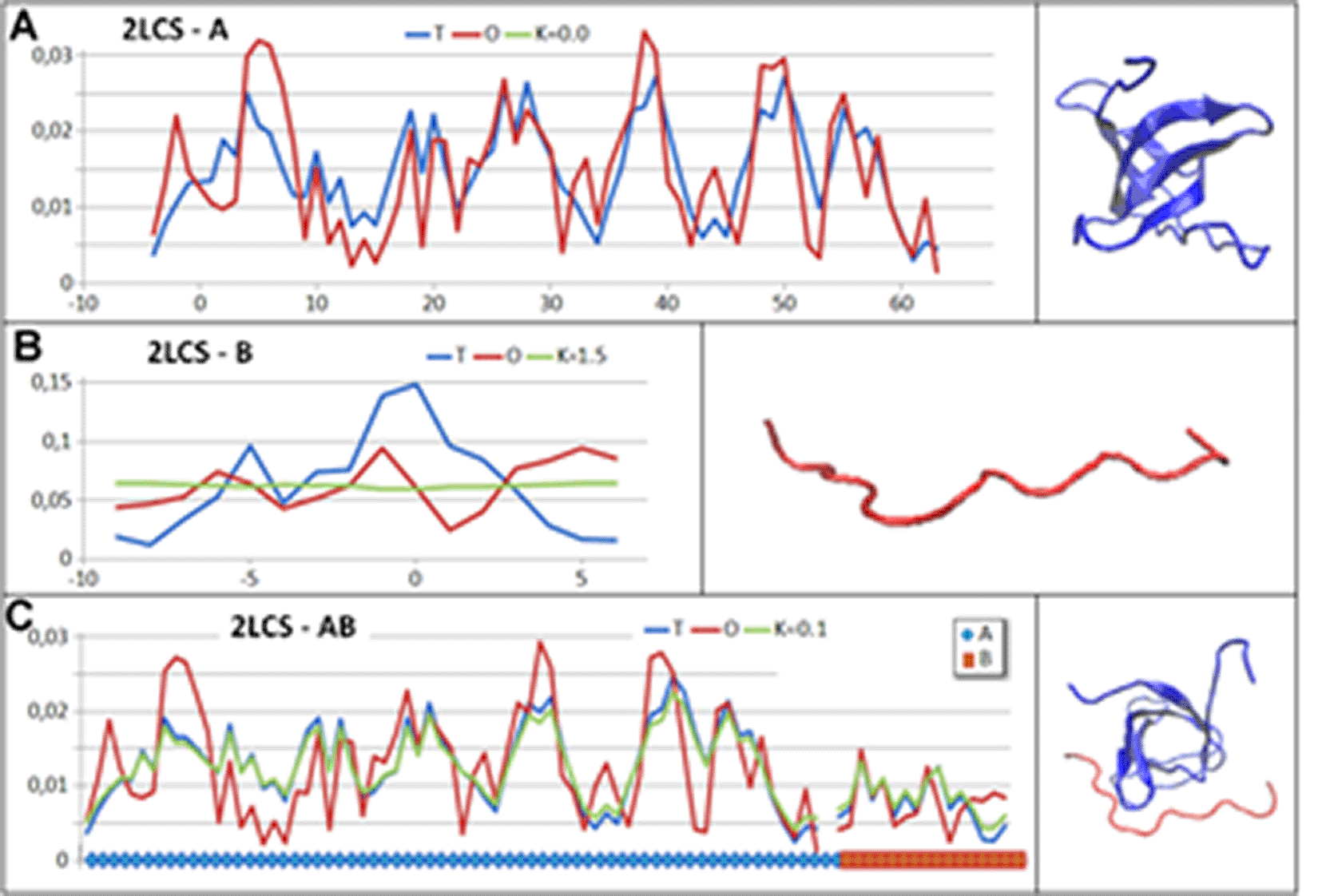
A – SH3 domain with 3D presentation.
B – polypeptide with 3D presentation.
C – dimer, sections corresponding to domain SH3 (blue) and peptide (red) are highlighted on the horizontal axis. The same colour scheme is used in the 3D presentation.
The alignment of the structure with the external force field is clearly visible in the form of a very low K value for the complex in question. This value is minimally different from K describing the SH3 status of the domain. This implies an alignment of the peptide also from the point of view of the hydrophobicity distribution of the dimer. Examples are given in Table. 4.
| CHAINS | RD | K | |
|---|---|---|---|
| 2LCS | A – SH3 | 0.330 | 0.0 |
| B – Peptide | 0.867 | 1.5 | |
| AB – complex | 0.361 | 0.1 | |
| 2VKN | A – SH3 | 0.365 | 0.2 |
| C – peptide | 0.634 | 0.4 | |
| AC – complex | 0.373 | 0.2 |
Analysis of the contribution of individual chains to the 2LCS dimer structure shows chain A status as a dimer component described with the values RD = 0.363 and K = 0.1 and for chain B with the values RD = 0.594 and K = 0.4. This set of parameter values expresses the relationship between the chains and the contribution to the construction of the common dimer structure. Dominant chain A shows a status comparable to that of the complex, while chain B expresses its role in determining the status of the complex with values significantly lower compared to its status as an individual structural unit. The value K = 0.4 can also be interpreted as determining the external force field provided by chain A, to which chain B has adapted. This example can be regarded as induced folding, which can also be expressed as folding under the influence of a local external force field. Chain A also acts as a chaperone imposing the folding of the chain according to the field generated by chain A.
In the case of the SH3 domain complex with the peptide from pbs2-kinase (PDB ID - 2VKN), the status of the peptide is determined by the value K = 0.4. This means that the SH3 domain provides an external force field directing folding to a lower degree than is the case in the example discussed above.
Another example of inducing folding is the folding of chain Alpha of haemoglobin assisted by AHSP, which essentially acts as a chaperone directing the folding of this chain. The role of AHSP is to prevent independent folding to the final form that the chain would probably have achieved without the directing factor.43 This chain treated as an individual structural unit (as available in PDB: 1W0B, 1W0A, 1W09) shows structuring with hydrophobicity distribution consistent with a micelle-like arrangement (Fig. 6.) (Table. 5). This means that the protein obtains its structure spontaneously in the aqueous environment.
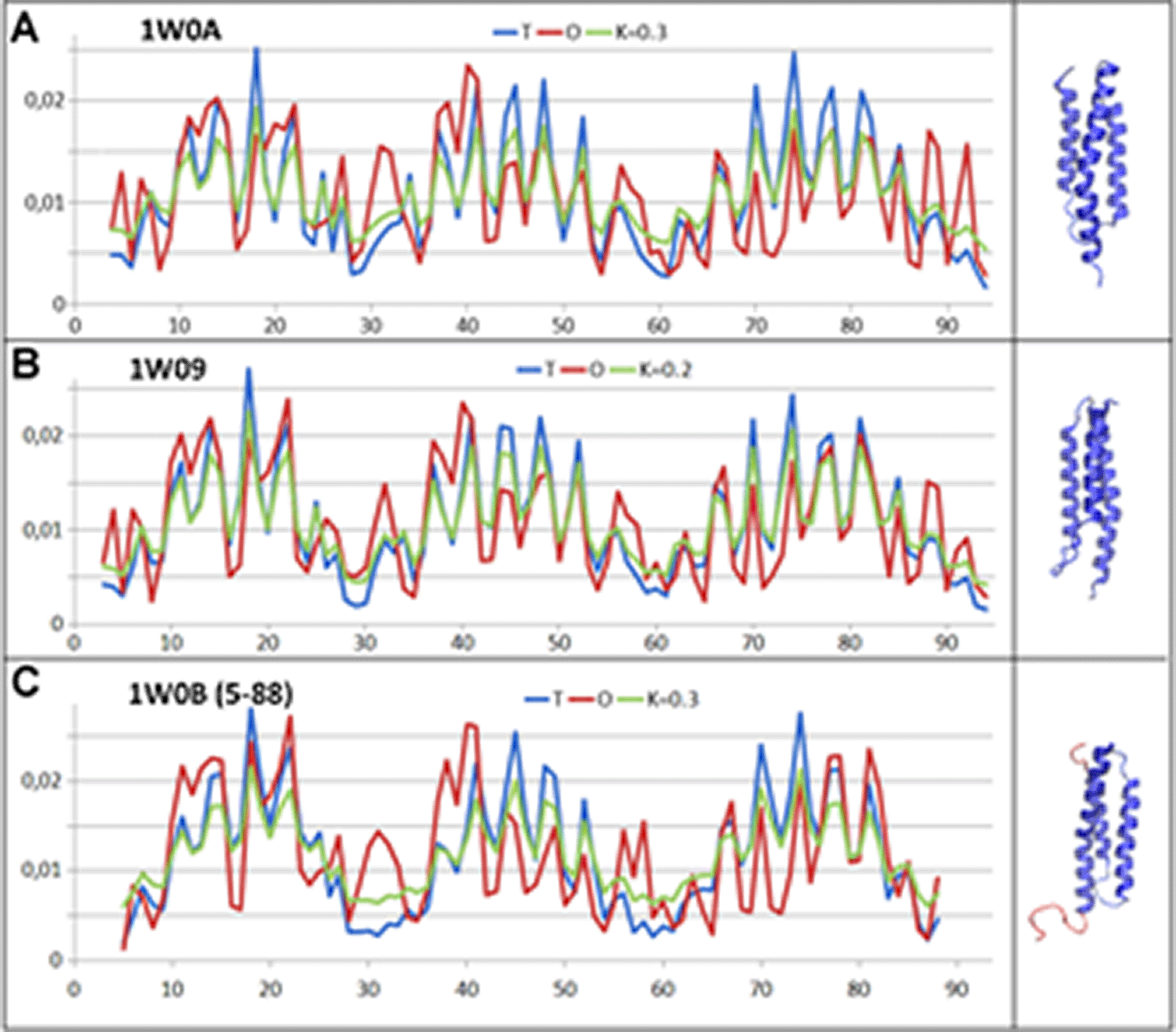
A – AHSP as available in PDB ID – 1W0A.
B – AHSP as available in PDB ID – 1W09.
C – AHSP as available in PDB ID 1W0B – N- and C-terminal fragments are highlighted (red) on the 3D presentation; these show a loose structure that is not part of the globular form of the protein. The profiles describe a fragment devoid of N- and C-terminal sections.
The protein with the code (PDB ID – 1Y0136) is the complex of AHSP with human Fe (II)-alphaHb. The low RD and K values for AHSP suggest the acquisition of structure by this protein exclusively under the influence of the aqueous environment (Fig. 7). The status of AHSP in complex with haemoglobin Alpha-chain is comparable to that shown as an individual structural unit.
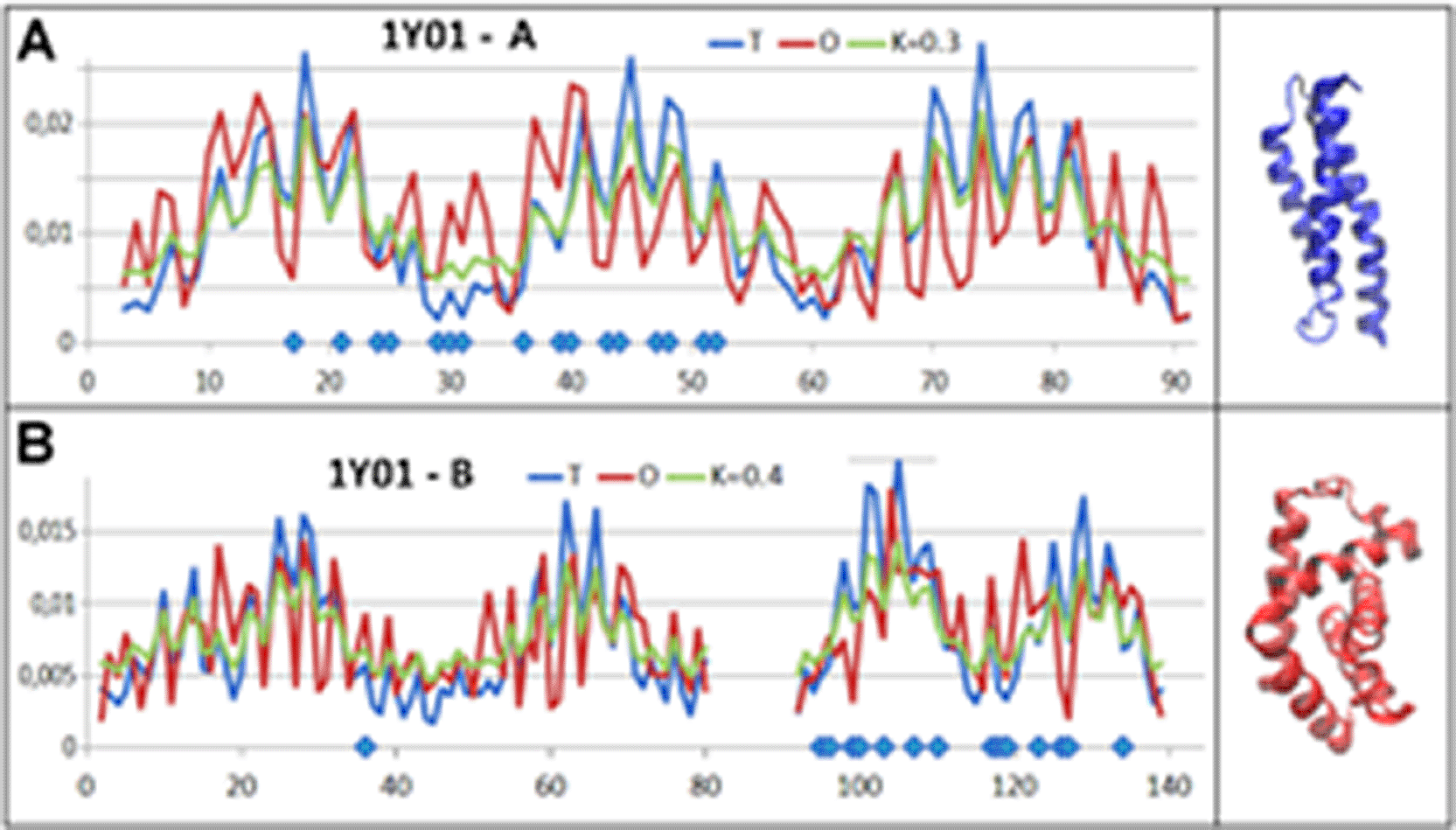
A – AHSP chain – positions of residues in contact with haemoglobin α-chain are highlighted on the horizontal axis.
B – haemoglobin Alpha chain – the positions of the residues in contact with the AHSP chain are highlighted on the horizontal axis.
The values of the RD parameters (Table. 6, Fig. 8) reveal the status of the residues involved in the interaction (P-P) and the part lacking the amino acids remaining in contact with the other chain. (No P-P) In the case of the complex, the status of the interface is favourable from the point of view of hydrophobicity distribution, which implies the participation of the interface in the construction of the common hydrophobic core. The remaining part (No P-P) devoid of the interface shows an increased RD value, indicating a significant role for the interface in the structure of the dimer. In contrast, the status of the residues involved in the interaction determined within the individual chains show elevated RD values for the residues comprising the interface. This entails preparing an exposure of hydrophobicity on the protein surface, and the exposure is used to form the complex.
The status (RD) of the interface in the dimer and the status of the residues within the structural unit (chains treated as individual structural units) included in the interface are given. P-P indicates the status of the residues comprising the interface. No P-P denotes the part of the analysed structural unit devoid of interacting residues.
| Protein | RD – P-P | RD – No P-P |
|---|---|---|
| Dimer: AHSP + haemoglobin Alpha chain AB | 0.484 | 0.529 |
| AHSP | 0.642 | 0.430 |
| Haemoglobin Alpha chain | 0.562 | 0.477 |
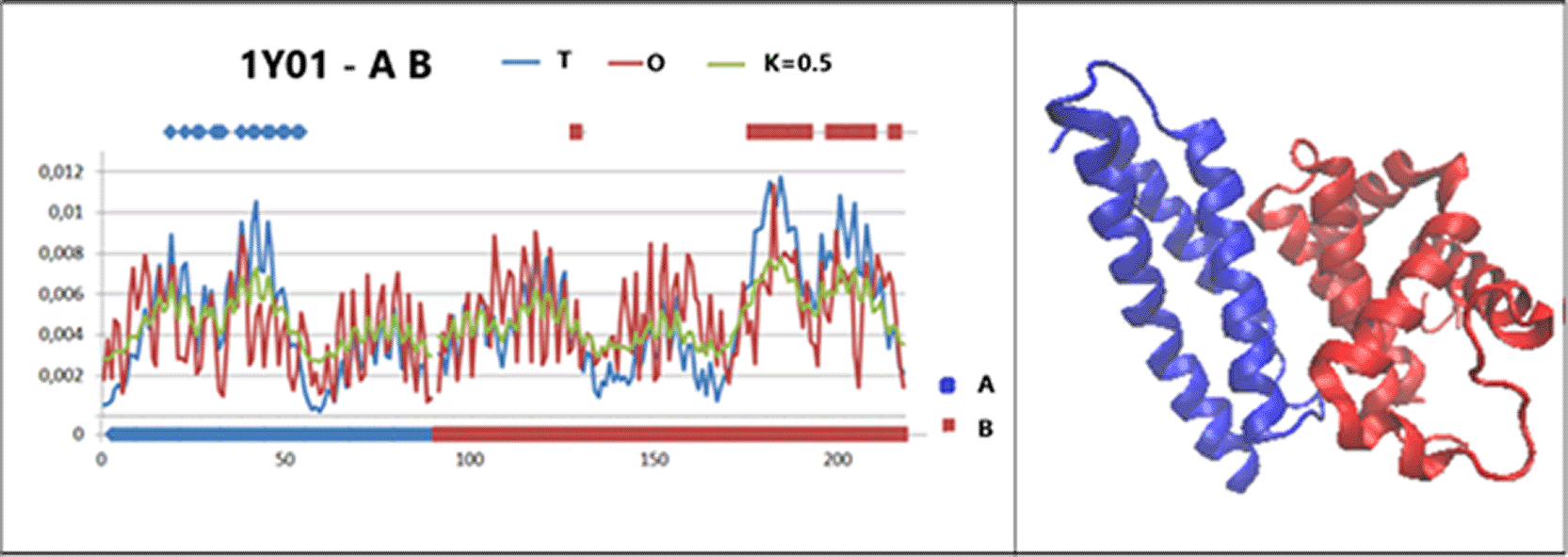
T (blue), O (red) and M (green) profiles for the value of K given in the legend together with a 3D presentation.
The horizontal axis shows the sections corresponding to: AHSD – blue, haemoglobin α-chain – red. Positions highlighted on the top axis – residues involved in inter-chain interaction.
The proteins and complexes discussed here show specific hydrophobicity distributions indicating the involvement of the aqueous environment as a factor directing the folding process. In the FOD-M model, this is indicated by the value of the K parameter, which introduces a modification to the record of the environment in which the structure is formed. For proteins showing K < 0.3, there is nothing surprising about their ease of folding and high folding rate in the experiment. For these proteins, it is also possible to reconstruct the structure by in silico methods if the parameterisation of the software used – including the force field in particular – is verified by the conditions of the aqueous environment. The dependence of the correctness of the models obtained by in silico methods for the given sequences in the CASP project was demonstrated in.44
Studies of protein structures based on the FOD-M model show a significant variation in the influence of the environment (external force field) on the folding process, which is evident in the final structure of a given protein (as available in PDB collections).
In addition to proteins belonging to the fast-folding group, the present work also discusses examples of induced folding – as defined in the cited work.37 Within the framework of the FOD-M model, induced folding is interpreted as a result of chemical components modifying the external force field of water, tailoring it to the specific environment exhibited by the analysed protein structure (chain B in the complex with SH3).
It is therefore postulated that the energy function optimisation procedure be a multi-criteria optimisation. In the case under discussion here, this function has the following form:
According to the FOD-M model, the achievement of a stable structure based on the funnel model is determined by the selection of an appropriate local energy minimum. This selection depends on the environmental influence, quantified by the value of parameter K (Fig. 9).47
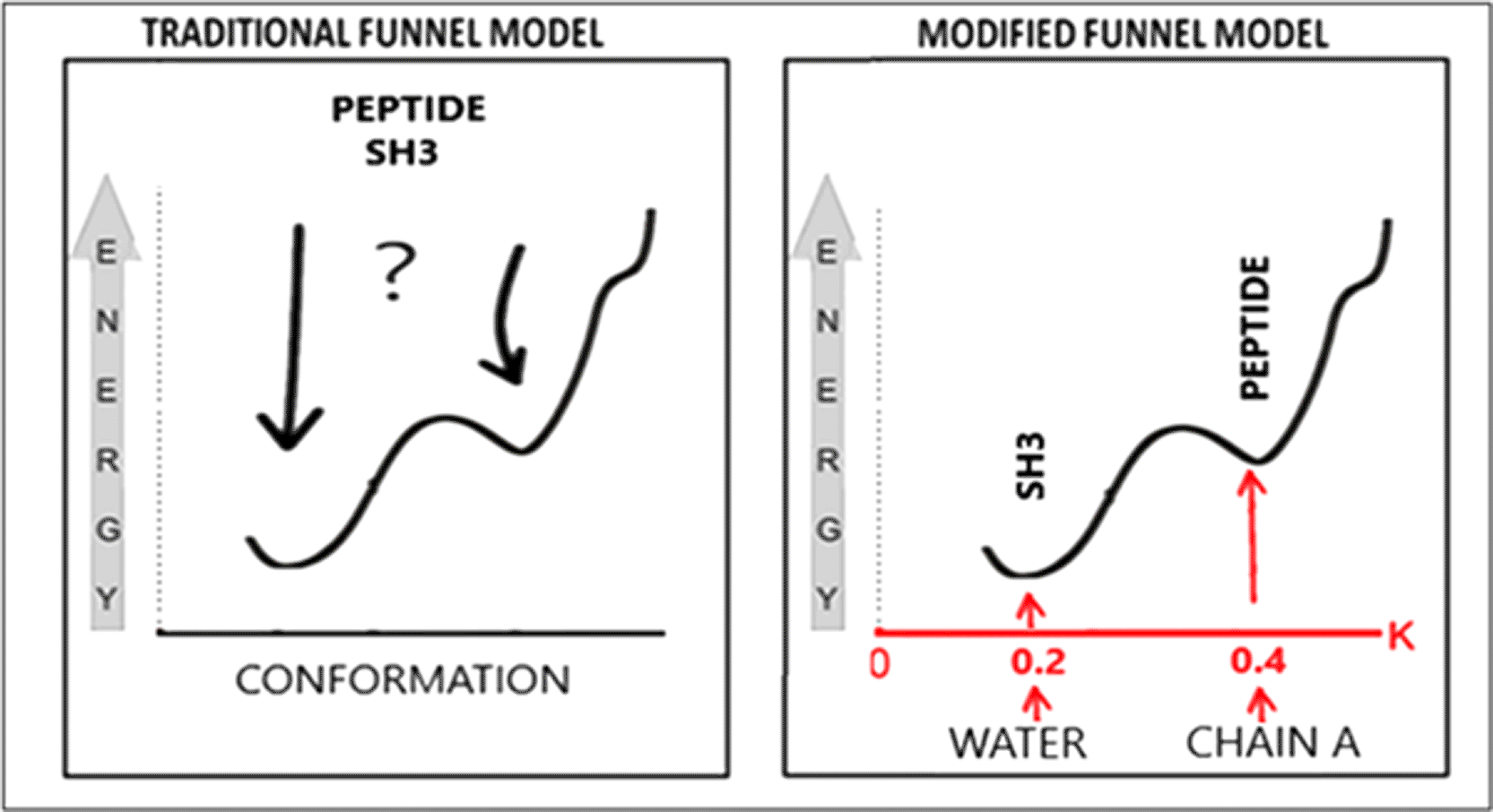
The example illustrates the components of the complex (SH3 and PDB ID peptide – 2VKN). Chain A in this dimer acts as a “chaperone” representing an external force field directing the structuring of the peptide in the complex.
The relatively high local energy minimum for chain A treated as an individual structural unit would probably have turned out to be somewhat lower without the presence of an external force field in the form of the SH3 domain. The chain could arguably achieve a lower energy structure in the aqueous environment (due to the hydrophobic core present). The presence of an external force field – in this case, the SH3 domain – stabilizes a higher-energy conformation of the protein (due to the weak hydrophobic core). Adjustment of chain A structuring in the complex leads to the energy state of the complex that provides stability.
In contrast, the Top7-CFr homo-dimer (PDB ID: 2GJH) exhibits a conformation – in both its monomeric and dimeric forms – that is fully consistent with the orientation imposed by the active aqueous environment. Hence the ease of both the folding of the monomeric unit itself and the formation and stability of the dimer.
The folding of proteins and the formation of complexes in the aqueous environment is mainly based on the adaptation of the hydrophobicity distribution to the environment. The evaluation of the structure of the dimer in question identifies this protein as a representative of fast-folding with parameter values based on the FOD-M model as representing a structure that constitutes the effect of the active participation of the aqueous environment in the folding process and dimer stabilisation.
What this means for this environment is the exposures of polar residues and the concentration of hydrophobic amino acids in the central part of the protein to form a hydrophobic core. A local deviation from the ideal O distribution towards the T distribution in the form of an exposure of hydrophobic residues on the surface is a signal of readiness for complex formation – in the case discussed in the present work – a dimer. A molecular dynamics simulation revealing structural changes in a protein or dimer takes into account non-binding interactions (electrostatic, vdW and torsional potential) and therefore only effects on EINT. A multi-object optimisation is proposed, where the expression for the energy status of a protein is a function of two functions: EINT (internal force field) and EEXT (external force field).
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)