Keywords
DNA barcodes, RNA barcodes, RNA tags, nanopore sequencing, direct RNA sequencing, long-read sequencing, high- throughput sequencing, multiplexing, demultiplexing, error tolerance, sample identification
This article is included in the Bioinformatics gateway.
DNA barcodes, RNA barcodes, RNA tags, nanopore sequencing, direct RNA sequencing, long-read sequencing, high- throughput sequencing, multiplexing, demultiplexing, error tolerance, sample identification
The emergence of long-read sequencing has revolutionized genomics and transcriptomics.1 Oxford Nanopore Technologies (ONT) platforms enable direct RNA sequencing without reverse transcription, preserving RNA modifications and full-length transcript information.2–4 However, these advantages come with distinct challenges: nanopore sequencing exhibits error rates of 5–15%, dominated by insertions and deletions rather than the substitution errors typical of short-read platforms.5,6
Sequence (DNA or RNA) barcodes or tags – short sequences used to identify samples, cells, or spatial locations in a pool of many DNA or RNA molecules – are essential for multiplexed sequencing.7,8 For short-read sequencing, barcodes of 8–12 bp (or nt) with minimum Hamming distances of 3–4 provide adequate error tolerance. The Hamming distance between two sequences of equal length counts the number of positions at which the corresponding bases differ; a minimum pairwise Hamming distance of d guarantees that up to ⌊(d − 1)/2⌋ substitution errors can be corrected. The Levenshtein (edit) distance – which additionally accounts for insertions and deletions – is a more comprehensive but computationally costlier measure of sequence dissimilarity. However, the indel-dominated error profile of nanopore sequencing demands longer barcodes with greater inter-sequence distances to maintain reliable sample identification.5,6 While sequencing barcodes (tags) are an essential molecular biology instrument, there have been unaddressed challenges in computationally designing robust longer tags with well-described, predictable behavior.
The computational challenge of barcode generation scales exponentially with length. Several tools exist for DNA barcode generation, each with distinct approaches and limitations ( Table 1). The widely-used DNABarcodes R package9,10 exhaustively enumerates all 4n possible sequences before filtering – 65,536 sequences at 8 bp, but 268 million at 14 bp. This approach fails at lengths above 12 bp due to memory exhaustion, precisely where long-read applications require robust barcodes. TagGD11 provides efficient demultiplexing but focuses on barcode assignment rather than de novo generation. BARCOSEL12 selects optimal subsets from existing barcode pools but cannot generate new sequences. FreeBarcodes13 generates indel-correcting barcodes but requires hours for large sets and targets specific code lengths. Edittag14 provides edit-distance-based design but with limited scalability. PRO15 takes a different approach designed specifically for nanopore applications: it uses a farthest-point sampling algorithm with probability divergence – a metric that models the sequencing error process more directly than edit distance – and includes built-in demultiplexing. However, PRO operates at a fixed barcode length matching the official ONT kit (24 bp) and targets maximizing barcode set size (up to 2,292 barcodes) rather than providing user-defined target counts, as well as requires the barcodes to be flanked by known adapter sequences. PRO offers no GC content filtering, homopolymer constraints, custom exclude sequences, graphical interface, or distance visualisation. None of these tools offer the combination of flexible barcode length (8–30 bp), sub-second generation, user-defined quality constraints, integrated demultiplexing, nor an accessible interface that nanopore researchers need to adapt barcode design to their specific error profiles.
Key capabilities of seven barcode generators are compared, including maximum practical barcode length, generation speed, interface availability, and constraint options. Symbols: ✓, supported; −, not supported or not applicable.
| Feature | TagGen | PRO | DNABarcodes | BARCOSEL | TagGD | FreeBarcodes | Edittag |
|---|---|---|---|---|---|---|---|
| Maximum practical length | 30 bp | 24 bp | ~12 bp | Any * | 16 bp | 20 bp | 12 bp |
| De novo generation | ✓ | ✓ | ✓ | – | – | ✓ | ✓ |
| Generation time (14 bp, 96 barcodes) | 54 ms | ~7 min | N/A † | – | – | Hours | N/A ‡ |
| Graphical interface | ✓ | – | – | Web | – | – | – |
| Command-line interface | ✓ | ✓ | ✓ | – | ✓ | ✓ | ✓ |
| Parallel processing | ✓ | – | – | – | – | – | – |
| Custom exclude sequences | ✓ | ✓ | ✓ | – | – | – | ✓ |
| GC content constraints | ✓ | – | ✓ | – | – | – | ✓ |
| Homopolymer filtering | ✓ | – | – | – | – | – | – |
| Distance visualization | ✓ | – | – | – | – | – | – |
| JSON config support | ✓ | – | – | – | – | – | – |
We developed TagGen to address this gap. By replacing exhaustive enumeration with Monte Carlo sampling, TagGen achieves linear complexity regardless of barcode length, enabling generation of 14–30 bp barcodes optimized for nanopore sequencing. Crucially, barcode generation alone is insufficient for a complete multiplexing workflow: once sequenced reads are obtained, each read must be assigned back to its source barcode – a process known as demultiplexing. Existing demultiplexers ( Table 2) typically require knowledge of flanking adapter sequences to anchor the barcode search, restricting them to standard kit-based library preparations. In contrast, TagGen includes an integrated anchor-free demultiplexer (taggen-demux) that locates barcodes at any position within a read – at the 5′ or 3′ end, or embedded mid-read – using a k-mer voting and banded edit-distance pipeline. This design enables demultiplexing of direct RNA sequencing reads, spatial transcriptomics libraries, and custom protocols where the barcode position is non-standard or variable. Position masks allow the user to restrict the search to the expected barcode region, minimising false positive matches while accommodating diverse library architectures. We validate these barcodes against realistic nanopore error profiles, demonstrating their resilience under conditions where traditional short barcodes fail. TagGen provides a complete barcoding pipeline: barcode design, simulation and quality validation, and post-sequencing demultiplexing. This integration removes the need to coordinate separately maintained tools and ensures that the distance parameters used during barcode generation are directly matched to the tolerances applied during demultiplexing. We describe the demultiplexer design and its systematic validation below.
Seven demultiplexers are compared across barcode flexibility, search strategy, distance metric, and maintenance status. Symbols: ✓, supported; ✗, discontinued; −, not supported.
| Feature | TagGen | minibar | PRO-demux | qcat | Dorado | Porechop | DeePlexiCon |
|---|---|---|---|---|---|---|---|
| Accepts user-defined barcodes | ✓ | ✓ | – | – | Limited † | ✓ | – |
| Tag location | Any | Any | 5′ | 5′ | 5′ or 3’ | 5′ or 3’ | 5′ |
| Adapter sequence required | – | ✓ | ✓ | ✓ | ✓ | ✓ | – |
| Distance metric | Edit | Edit | Prob. div. | Edit | Edit | Edit | Neural net |
| Configurable mismatch threshold | ✓ | ✓ | – | – | – | – | – |
| Indel-aware matching | ✓ | ✓ | ✓ | Partial | ✓ | Partial | ✓ |
| Real-time/live support | – | – | – | – | ✓ | – | – |
| GPU acceleration | – | – | – | – | ✓ | – | ✓ |
| Active maintenance | ✓ | ✓ | ✓ | ✗ ‡ | ✓ | ✗ ‡ | Limited |
| Graphical interface | ✓ | – | – | – | – | – | – |
| Command-line interface | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
TagGen implements a two-phase algorithm fundamentally different from exhaustive enumeration ( Figure 1).
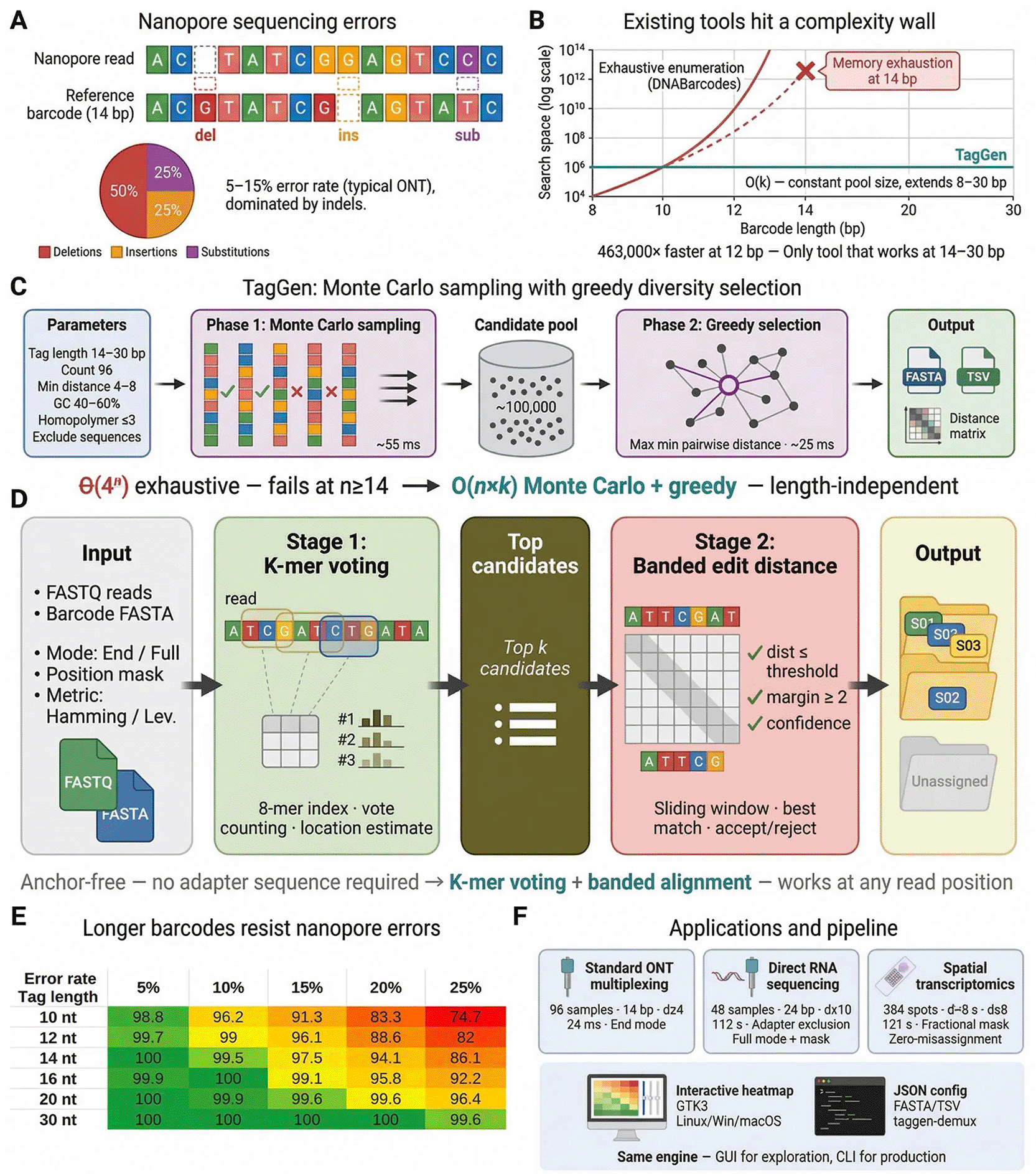
(A) Nanopore sequencing errors are dominated by deletions (50%), insertions (25%), and substitutions (25%) at 5–15% total error rates, requiring longer barcodes than short-read platforms. (B) Existing tools enumerate all 4n sequences exhaustively, causing memory exhaustion at ≥14 bp (268 million sequences). TagGen’s Monte Carlo sampling operates at constant complexity O(k) regardless of length. (C) TagGen’s two-phase generation algorithm: Phase 1 generates ~100,000 random candidates validated against user constraints in parallel; Phase 2 applies greedy diversity selection maximising minimum pairwise Hamming or Levenshtein distance. (D) TagGen’s integrated demultiplexer: a two-stage anchor-free pipeline that assigns reads to source barcodes without requiring flanking adapter sequences. Stage 1 uses k-mer voting to rapidly identify candidate barcodes and estimate their location; Stage 2 performs banded edit-distance alignment to confirm the best match against configurable acceptance criteria. (E) Validation under a nanopore error model demonstrates that 30 bp barcodes (d ≥ 8) maintain 100% correct assignment at 20% error rate, while 10 bp barcodes degrade to 83%. Barcodes ≥14 bp achieve >97% resolution at typical ONT error rates (10–15%). (F) TagGen addresses sample multiplexing, direct RNA sequencing, and spatial transcriptomics through an integrated GUI and CLI pipeline with built-in demultiplexing.
Phase 1: Monte Carlo Candidate Generation. Rather than enumerating all 4n sequences, TagGen generates random candidates validated in real-time against user-specified constraints (GC content, homopolymer limits, exclude sequences). Generation proceeds in parallel across CPU cores and terminates upon collecting sufficient valid candidates (typically ~1,250 candidates in ~55 ms).
Phase 2: Greedy Diversity Selection. From the candidate pool, TagGen iteratively selects sequences maximizing minimum Hamming or Levenshtein distance to already-selected barcodes, producing a maximally diverse final set (typically 96 barcodes in ~25 ms additional time).
The following pseudocode formalizes TagGen’s two-phase approach.
Input: length, targetCandidates, gcMin, gcMax, homopolymerLimit, excludeSeqs
Output: candidatePool (set of valid barcode candidates)
1. Initialize candidatePool ← ∅
2. Initialize lock for thread-safe access
3. parallel for each CPU core do:
4. while|candidatePool|< targetCandidates do:
5. seq ← generateRandomSequence (length) //uniform random A,C,G,T
6. //Validate constraints
7. if gcContent (seq) < gcMin or gcContent (seq) > gcMax then
8. continue
9. if maxHomopolymerRun (seq) > homopolymerLimit then
10. continue
11. if seq ∈ excludeSeqs then
12. continue
13. //Thread-safe addition
14. acquire (lock)
15. candidatePool ← candidatePool ∪ {seq}
16. release (lock)
17. end while
18. end parallel
19. return candidatePool
Input: candidatePool, targetCount, minDistance
Output: selectedBarcodes (maximally diverse subset)
1. Initialize selectedBarcodes ← ∅
2. Initialize distanceMatrix [I,j] for all pairs in candidatePool//distance can be hamming (default, fastest) or Levenshtein (slower, indels considered)
3. //Select first barcode (arbitrary or random)
4. selectedBarcodes ← selectedBarcodes ∪ {candidatePool[0]}
5. while|selectedBarcodes|< targetCount do:
6. bestCandidate ← null
7. bestMinDist ← -1
8. for each candidate in candidatePool \ selectedBarcodes do:
9. //Find minimum distance to any selected barcode
10. minDist ← min {distanceMatrix [candidate, s] : s ∈ selectedBarcodes} : s ∈ selectedBarcodes}
11. if minDist > bestMinDist then
12. bestMinDist ← minDist
13. bestCandidate ← candidate
14. end for
15. if bestMinDist < minDistance then
16. break//Cannot achieve required minimum distance
17. selectedBarcodes ← selectedBarcodes ∪ {bestCandidate}
18. end while
19. return selectedBarcodes
The algorithmic approaches differ fundamentally in complexity:
• Exhaustive enumeration: O(4n) where n = barcode length
• Monte Carlo sampling: O(k) where k = candidates generated (typically 10,000–100,000)
For 14 bp barcodes, this represents a 2,680-fold reduction in operations (100,000 vs. 268 million), translating to milliseconds versus memory exhaustion. Generation time is largely independent of candidate pool size for a fixed target count (Extended Data Figure 1).
TagGen includes a built-in demultiplexer (also standalone as taggen-demux) that assigns ONT FASTQ reads to their source barcodes. It is available both as a command-line subcommand (taggen --demux) and as a dedicated tab in the graphical interface.
Each read is processed by a two-stage pipeline. In the first stage, all 8-mers in the read’s search region are looked up against a pre-built index that maps each 8-mer to the set of barcodes containing it, and candidate barcodes are ranked by hit count (k-mer voting). The top candidate’s hit positions are averaged to estimate the barcode location within the read. In the second stage, a banded edit distance computation slides the candidate barcode across a window of (tag length + 15) bp centred on the estimated location, yielding the minimum edit distance to the read. The best-matching barcode is accepted if three criteria are met: (i) the edit distance does not exceed the automatically derived maximum (adaptive rule: tag_length/5 for tags shorter than 20 bp, tag_length/4 for 20–29 bp, tag_length/3 for ≥30 bp; overridable via --max-dist); (ii) the margin between the best and second-best candidate distance is at least 2 (ambiguity guard); and (iii) a confidence score derived from the edit distance exceeds a user-configurable threshold (default 0). Reads failing any criterion are written to an “unassigned” output file, annotated with the reason (no_match, ambiguous, or low_confidence). This conservative strategy ensures that uncertain reads are withheld rather than wrongly assigned.
Both Hamming distance (substitutions only) and Levenshtein edit distance (substitutions, insertions, deletions) are supported and applied consistently between barcode generation and demultiplexing. Because ONT sequencing produces a characteristic error profile with a significant proportion of insertion and deletion errors – particularly at homopolymer runs – Levenshtein distance is recommended for most ONT workflows. Hamming distance remains available for platforms with predominantly substitution errors or when computational speed is a priority.
Two search modes accommodate different library preparation strategies, as follows.
End mode (default): the demultiplexer searches a window at each end of the read and its reverse complement, corresponding to the standard dual-ended library structure [barcode][adapter][insert] [RC (adapter)][RC (barcode)]. This is the appropriate mode for ligation-based ONT DNA libraries.
Full mode: the entire read is scanned for the barcode, intended for protocols where the barcode may appear at an internal position (e.g., direct RNA sequencing with a mid-read adapter ligation, or custom in-read tagging schemes). In full mode, users can specify one or more position masks to restrict the search to the expected barcode region, reducing false positive k-mer seeds from flanking sequence. Masks may be expressed as absolute positions (start:end in bp), end-relative offsets (5p:N for the first N bp from the 5′ end; 3p:N for the last N bp from the 3′ end), or as read-length fractions (f_start:f_end,resolved per read). Multiple zones are merged and the union is searched.
Three trimming modes are available: none (reads written unmodified), ends (barcode trimmed from the matched end), and all (barcodes trimmed from both ends). FASTQ headers of assigned reads are annotated with the barcode name, edit distance, and confidence score. Assigned reads are written to per-sample subdirectories; unassigned reads to a separate subdirectory. Gzip compressed input is supported and processed in streaming batches to maintain constant memory usage. A per-sample statistics TSV file records the number of reads assigned per barcode, expected read count, and mean Q-score, enabling rapid quality assessment. In the GUI, these statistics are displayed as four interactive charts: barcode match position distribution, per sample assignment counts, edit distance and confidence distributions, and per-sample Q-score profiles.
When raw POD5 signal files are present alongside FASTQ reads, TagGen can invoke the pod5 subset tool to partition signal files by sample, enabling downstream signal-level analyses (e.g., modified base calling) on a per-sample basis.
TagGen is implemented in D (dlang.org), a systems programming language combining high-level expressiveness with native code performance. The application is compiled using the LDC2 compiler (LLVM-based D compiler) with release optimizations enabled.
The software architecture comprises six primary modules ( Figure 1):
1. Candidate Generation Module: Implements parallel Monte Carlo sampling using D’s std.parallelism library. Each worker thread independently generates random nucleotide sequences and validates them against user constraints in real-time. Valid candidates are collected into a thread-safe pool using atomic operations (Extended Data Figure 2). The module employs the Mersenne Twister pseudorandom number generator for sequence generation.
2. Diversity Selection Module: Implements greedy subset selection with Hamming or Levenshtein distance calculation. The algorithm maintains a distance matrix and iteratively selects the candidate maximizing minimum distance to all previously selected barcodes. Distance calculations use optimized bitwise operations for performance.
3. GUI Module: Built using GtkD bindings to the GTK3 toolkit, providing a cross-platform graphical interface. The interface includes real-time parameter validation, progress indication, and interactive heatmap visualization of pairwise barcode distances using Cairo graphics.
4. CLI Module: Provides complete command-line functionality for scriptable pipeline integration. Supports all generation parameters, JSON configuration files for reproducible workflows, multiple output formats, and verbose progress reporting.
5. Export Module: Generates output in FASTA format (for direct use in sequencing pipelines), TSV format (for spreadsheet analysis), and optional distance matrix export, with configurable sequence headers and metadata.
6. Demultiplexer Module: Implements a two-stage read assignment pipeline. The first stage uses a k-mer index (8-mer vocabulary) for rapid candidate identification via vote counting; the second stage performs banded edit-distance alignment to confirm the best match. The module supports end-mode and full-mode search with configurable position masks, adaptive acceptance thresholds, ambiguity detection, and three tag-trimming modes. POD5 signal file co-demultiplexing is supported via the pod5 subset command-line tool.
Minimum System Requirements:
• Operating System: Linux (Arch, Ubuntu 18.04+, Debian 10+), Windows 10+, or macOS 10.14+
• Processor: Any x86–64 CPU; multi-core processors recommended for parallel generation
• Memory: 512 MB RAM minimum; 2 GB recommended for large candidate pools
• Disk Space: 50 MB for installation
• Display: 1024 × 768 minimum resolution for GUI (not required for CLI operation)
Installation: Pre-compiled binaries are available for Linux and Windows from the GitHub releases page. Linux users may also compile from source using the DUB package manager:
dub build --compiler = ldc2 --build = release
GUI Workflow:
1. Launch TagGen GUI:. /taggen
2. Configure barcode parameters:
Tag length (8–30 bp recommended).
Target barcode count (e.g., 96 for standard plates).
Minimum Hamming distance (3–8 depending on error tolerance required)
3. Set quality constraints:
GC content bounds (default: 30–70%).
Maximum homopolymer length (default: 3)
4. Optionally load exclude sequences (FASTA format) to avoid similarity to adapters or primers
5. Click “Generate” to produce barcodes
6. Review pairwise distance heatmap for quality verification
7. Export to FASTA or TSV format
Demultiplexing workflow (Demultiplex tab):
8. Switch to the Demultiplex tab
9. Load barcode FASTA file (generated above or user-supplied)
10. Select FASTQ read file(s) for demultiplexing
11. Choose search mode: end (for standard libraries) or full (for mid-read barcodes)
12. Optionally configure position mask, acceptance threshold, and trim mode
13. Click “Demultiplex” to assign reads to barcodes
14. Review interactive result charts: per-sample assignment counts, barcode match positions, edit distance distributions, and per-sample Q-score profiles
15. Demultiplexed reads are written to per-sample subdirectories; unassigned reads to a separate directory
Command-Line Usage:
TagGen provides a comprehensive CLI for automated pipelines. Barcode generation and demultiplexing examples are shown below:
# Generate 96 barcodes with default parameters taggen -n 96 -l 14 -d 4 -o my_barcodes
# Generate with custom GC constraints and exclude sequences taggen -n 96 -l 16 --minGc 40 --maxGc 60 -f adapters.fasta -v
# Generate using JSON configuration file taggen -c experiment_config.json
# Generate large pool with matrix output taggen -n 384 -l 20 -d 6 --metric levenshtein -m --heatmap -v
# Demultiplex reads in end mode (standard dual-ended libraries) taggen --demux --tags my_barcodes.fasta --reads reads.fastq --mode end --trim-mode ends
# Demultiplex with full-mode search and 5′ position mask taggen --demux --tags my_barcodes.fasta --reads reads.fastq --mode full --position-mask 5p:60
# Demultiplex mid-read barcodes with fractional mask and strict threshold taggen --demux --tags my_barcodes.fasta --reads reads.fastq --mode full --position-mask 0.05:0.20 --max-dist 4
Full CLI options are available via taggen –help.
To validate barcode resilience under realistic long-read conditions, we implemented a nanopore error simulator based on published characterizations.5,6,18 Deletions: 50% of errors (most frequent in nanopore sequencing). Insertions: 25% of errors (random nucleotide insertion). Substitutions: 25% of errors (random nucleotide replacement). We tested error rates from 5% (high-quality R10.4 data) to 25% (stress test beyond typical conditions), encompassing typical ONT performance (10–15%) and degraded sample scenarios (20%). For each condition, 1,000 reads were simulated per barcode, with identification via minimum Levenshtein distance to the original barcode set.
Performance comparisons were conducted on a workstation with a 12-core AMD processor running Arch Linux. TagGen (v1.2.0) barcode generation runtime was compared against DNABarcodes (v1.20.0) from Bioconductor.10 Both tools were configured to generate 96 barcodes with equivalent constraint parameters (GC content 25–75%, minimum Hamming distance as specified). Timing measurements excluded I/O operations to focus on algorithmic performance. Each TagGen configuration was tested in triplicate; reported values are means ( Table 3).
Performance comparison between TagGen and DNABarcodes. Wall-clock generation times (mean of three replicates) for 96 barcodes at increasing lengths. All runs used Hamming distance and GC 25–75% on a 12-core AMD Linux workstation.
The practical sequencing utility of TagGen-generated barcodes was evaluated by simulating Oxford Nanopore reads with Badread v0.4.119 and measuring demultiplexing accuracy against ground truth. Reads were simulated using the nanopore2023 error model across read identity levels of 80–95% (corresponding to approximately 5–20% per-base error rates) spanning the range from early ONT chemistries to most recent high-accuracy data. Two demultiplexers were applied to the same simulated reads: taggen-demux (integrated, using Levenshtein distance) and minibar v0.25 (an established third-party ONT demultiplexer).20 Results are reported as accuracy (percentage of reads correctly assigned to their source barcode) and error rate (reads assigned to a wrong barcode); reads failing matching criteria are classified as unassigned rather than wrong.
Several other tools in this space were not included in the comparison. Qcat,16 ONT’s open-source demultiplexer, is designed exclusively for ONT’s predefined native barcode kits and does not accept arbitrary user-defined sequences; similarly, the demultiplexing module integrated into ONT’s Dorado basecaller21 is restricted to native barcode catalogues. Porechop17 has been discontinued and does not support user-defined barcodes. PRO15 is publicly available but a direct comparison remains impractical: PRO optimises for maximum barcode set size at a fixed length rather than accepting user-defined target counts and length ranges, and offers no GC content filtering, homopolymer constraints, or custom exclude sequences. These different design goals make a like-for-like accuracy comparison uninformative.
Demultiplexing accuracy was evaluated across 154 parameter combinations in five systematic test phases using the same Badread simulation setup described above. Tag sets were generated with TagGen using a pool of 10,000 candidates, GC content 30–70%, maximum homopolymer 3 bp, and error tolerance t = 2. A hundred reads were simulated per barcode per run; ground-truth assignments were extracted from Badread read headers. Demultiplexing was performed with taggen-demux using an adaptive acceptance threshold (autoMaxDist: tag_length/5 for tags shorter than 20 bp, tag_length/4 for 20–29 bp, and tag_length/3 for tags of 30 bp or longer) unless otherwise noted. Three accuracy metrics were recorded for each condition: the fraction of reads correctly assigned to the originating sample (accuracy), the fraction assigned to an incorrect sample (misassignment error), and the fraction rejected as unassigned.
We benchmarked TagGen against DNABarcodes across configurations spanning short-read (8–12 bp) and long-read (14–30 bp) applications ( Figure 2, Table 3).
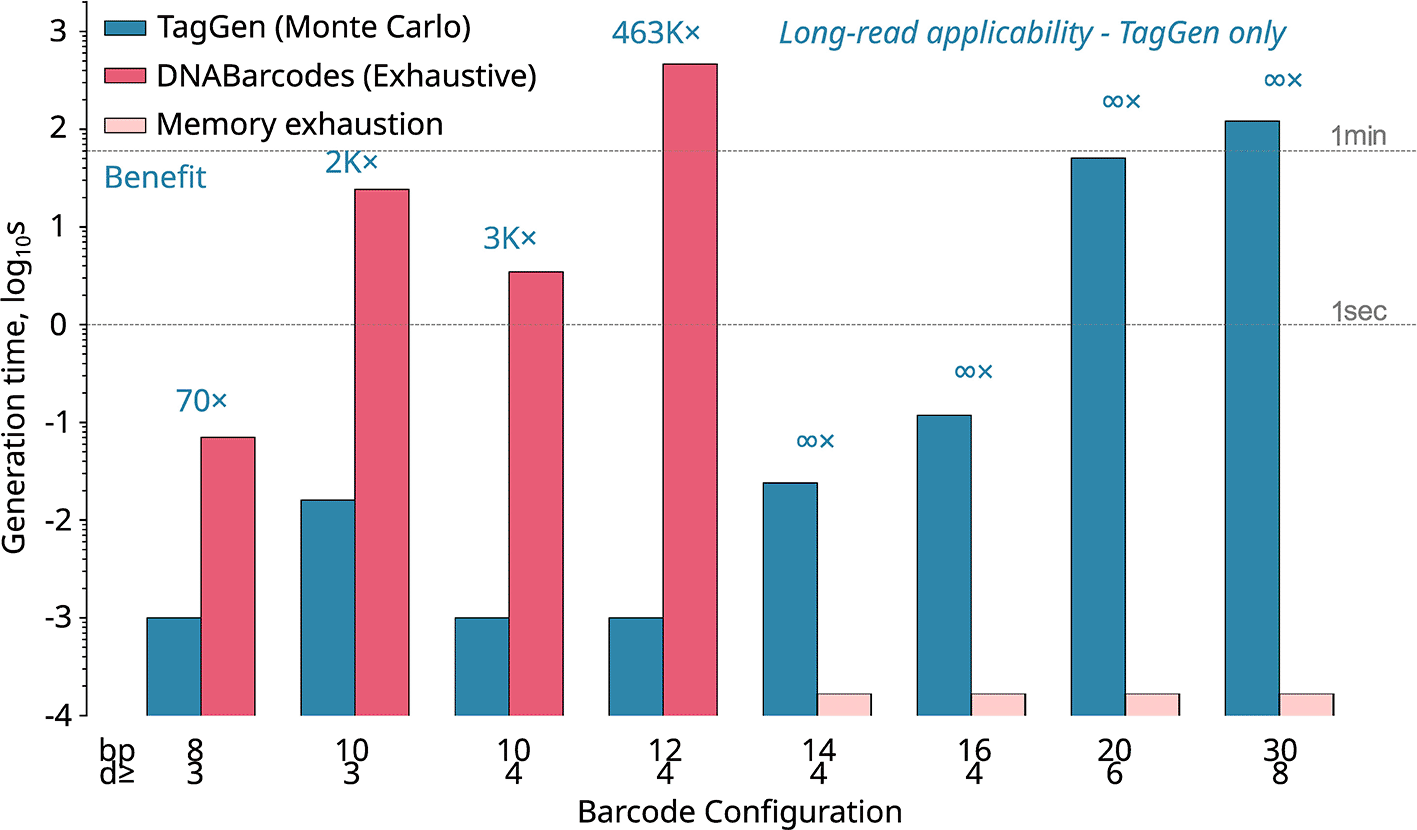
Generation times (log scale) across barcode lengths 8–30 bp. DNABarcodes (orange) fails at ≥14 bp due to memory exhaustion when attempting to enumerate 268+ million sequences. TagGen (blue) succeeds at all lengths with sub-second generation times for typical applications (14–20 bp).
All benchmarks: 96 target barcodes, Hamming distance, GC 30-70%. Wall-clock times averaged over 3 replicates, Linux. DNABarcodes memory exhaustion occurs when attempting to enumerate 4n sequences (268 million at 14 bp).
At comparable configurations, TagGen is able to generate longer barcodes (14–30 bp range) with generation times below 50 ms for all tested lengths (Extended Data Figure 3), while the exhaustive enumeration strategy of DNABarcodes fails entirely due to memory requirements exceeding available RAM when attempting to store 268+ million candidate sequences.
Using a literature-based error model on the tags directly, we tested barcode resolution across lengths and error rates, showing that the generated tags are robust to most error rates, in particular longer barcodes ( Table 4).
Percentage of reads correctly assigned to the originating barcode across five error rates (5–25%) for six barcode lengths. Error model: 50% deletions, 25% insertions, 25% substitutions; N = 1,000 simulated reads per barcode per condition.
Values indicate percentage of reads correctly assigned to original barcode. Error model: 50% deletions, 25% insertions, 25% substitutions based on published ONT characterizations. N = 1,000 simulated reads per barcode per condition.
The results reveal a striking pattern. At typical nanopore error rates (10–15%), traditional 10 bp barcodes show degradation (91–96% correct), while barcodes ≥14 bp maintain >97% resolution. Under challenging conditions (20% error) – representing degraded RNA or difficult sequences – the contrast is dramatic: 10 bp barcodes achieve only 83.3% correct assignment (16.7% misassignment), while 30 bp barcodes maintain 100% correct assignment. Even at the extreme 25% error stress test, 30 bp barcodes maintain 99.6% accuracy while 10 bp barcodes fall to 74.7%.
To benchmark taggen-demux in a standard dual-ended library context, we compared its performance against minibar (v0.25), a widely used primer-anchored demultiplexer for ONT data. Simulated reads were generated with a dual-ended structure ([barcode [adapter][insert] [RC (adapter)][RC (barcode)]) using the same TagGen-generated Levenshtein barcodes (n = 96, l = 14/20/30 bp, d = 4/8/8) and Badread simulator used for the rest of the benchmarks. Both tools were given the same barcode FASTA; minibar was additionally provided the flanking adapter sequence, which taggen-demux does not use. The two tools differ fundamentally in their matching strategy. Taggen-demux performs an anchor-free alignment: it searches a window at the read end and computes the edit distance between each reference barcode and the raw read sequence. It therefore operates without any knowledge of where the barcode ends and the adapter begins. Minibar uses a two-step primer-anchored approach: it first locates the adapter by alignment, then extracts the bases immediately upstream as the barcode, and finally compares only those extracted bases against the reference set. When sequencing indels shift the barcode region within the read, primer-anchored extraction isolates the barcode cleanly; the anchor-free approach must absorb those indels within its edit-distance threshold. At high read identity (≥90%), minibar assigned more reads correctly than taggen-demux for 20 and 30 bp barcodes (95–100% vs 78–97%), with near-zero misassignment in both cases ( Figure 3). At 80% read identity, where an average 30 bp barcode carries approximately six sequencing errors, minibar still achieved 88% accuracy for 30 bp barcodes, compared to 59% for taggen-demux. For short barcodes (14 bp) at low identity (80–85%), the two tools performed comparably (17–42%), as the adapter itself becomes too corrupted for reliable anchoring and the tight acceptance threshold (2 edit operations) limits both approaches equally. Taggen-demux consistently produced a small misassignment rate (0.05–1.4%), while minibar produced near-zero misassignment across all conditions. The higher misassignment for taggen-demux at l = 20 bp and low identity reflects the more liberal acceptance threshold (edit_dist = 5 out of 20 bp = 25% of tag length) combined with the absence of primer-assisted barcode extraction. These results confirm that when flanking adapter sequences are known and library structure is fixed, primer-anchored tools such as minibar extract barcodes with higher sensitivity. Taggen-demux is not designed to compete in this scenario; its intended contribution lies in settings where primer sequences are unavailable, variable in position, or absent by design – including direct RNA sequencing, spatial transcriptomics capture plates, and custom library structures in which the barcode is not flanked by a consistent adapter. These scenarios, which minibar cannot address, are evaluated in the following sections.
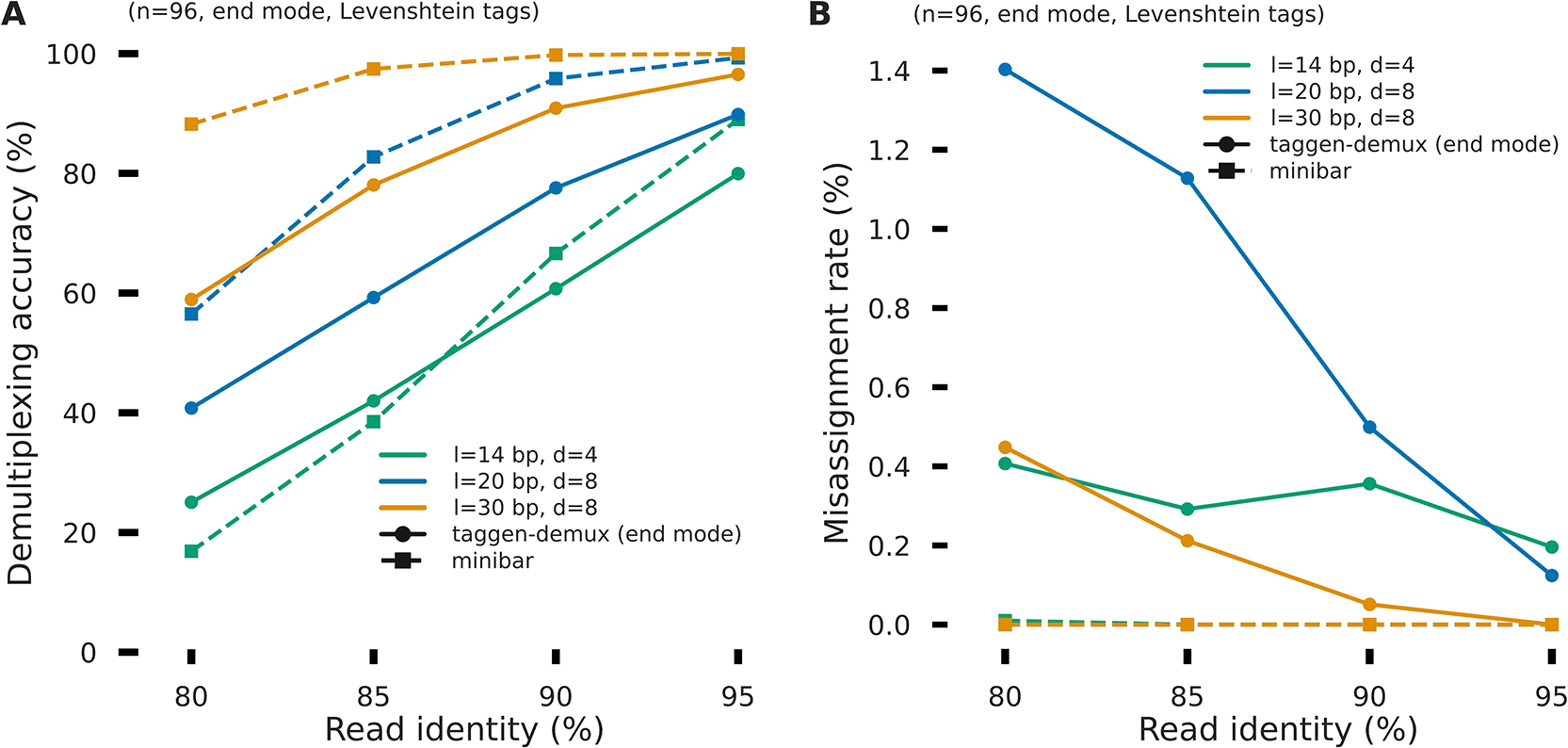
Both tools were applied to the same simulated dual-ended ONT reads ([barcode][adapter][insert] [RC (adapter)][RC (barcode)], 100 reads per barcode, n = 96, Badread nanopore2023 error model). taggen-demux operated in end mode without primer information; minibar additionally received the flanking adapter sequence as a positional anchor. (A) Demultiplexing accuracy and (B) misassignment rate as a function of simulated read identity, for three barcode lengths and minimum pairwise Levenshtein distances (l = 14 bp d = 4, l = 20 bp d = 8, l = 30 bp d = 8). Solid lines, taggen-demux; dashed lines, minibar. Minibar achieves higher assignment rates at medium-to-high read identity by leveraging the primer anchor, whereas taggen-demux is designed for library configurations in which no such anchor is available.
The key advantage of longer barcodes becomes evident under simulated nanopore errors ( Table 4, Figure 4). End-mode demultiplexing accuracy increased monotonically with both tag length and read identity ( Figure 4A). At 95% read identity – representative of latest ONT chemistry – accuracy reached 84.7%, 92.2%, 93.3%, and 97.5% for tag lengths of 14, 20, 24, and 30 bp, respectively, with near-zero misassignment (<0.03%) in all cases (Extended Data Figure 4). At 85% identity, which corresponds to older R9.4.1 flow cells or low-complexity regions, accuracy ranged from 42.2% (14 bp) to 77.1% (30 bp). Reads that could not be confidently assigned were rejected as unassigned rather than being misassigned, preserving the integrity of all assigned reads. We therefore recommend 30 bp Levenshtein-generated tags for experiments where read quality is expected to be moderate (≤90% identity).
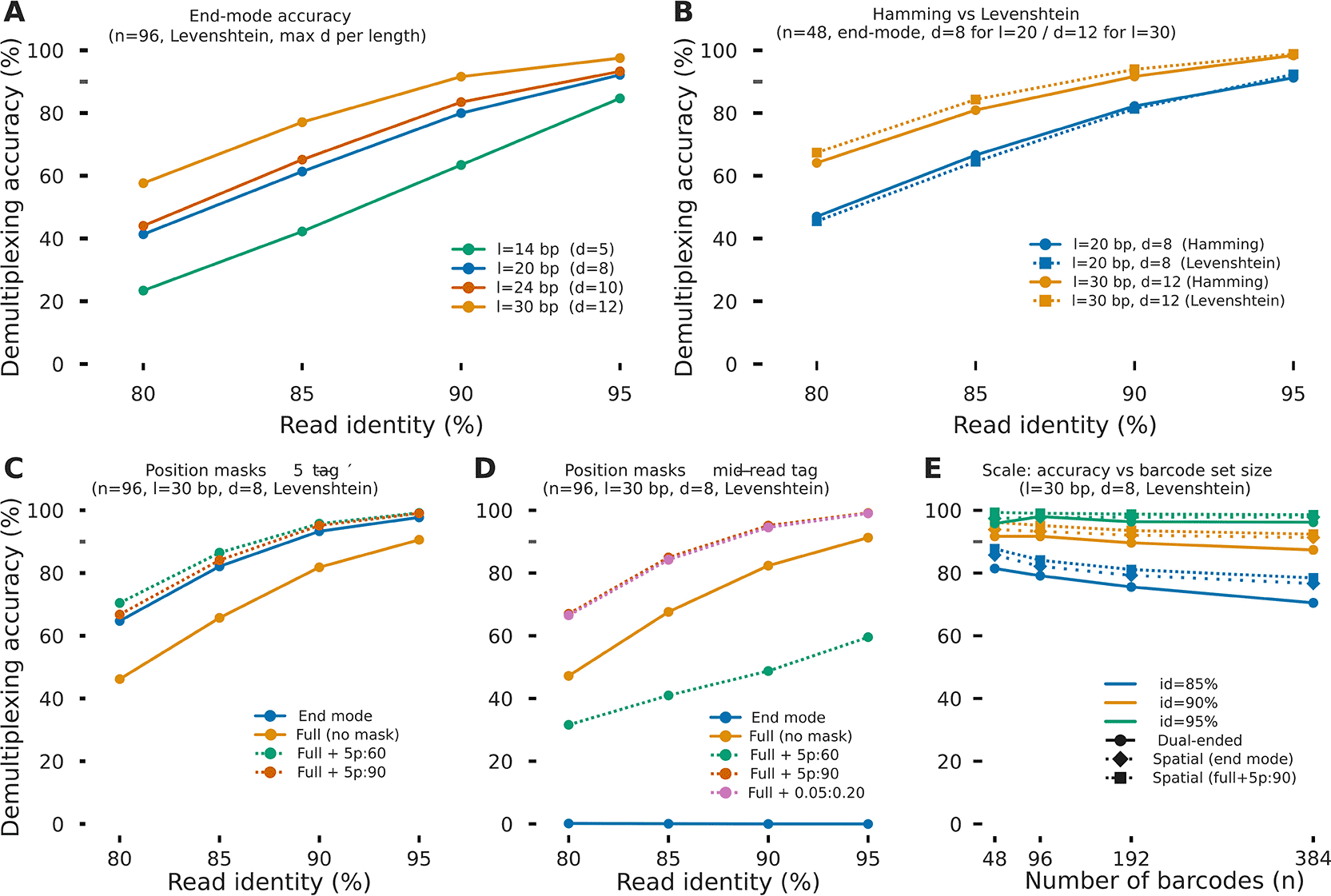
All simulations used Badread (nanopore2023 error model) at four read identity levels (80–95%) representing the range from older R9.4.1 to current R10.4.1/Kit14 chemistry. (A) End-mode demultiplexing accuracy as a function of read identity for four barcode lengths (14, 20, 24, and 30 bp) generated with Levenshtein distance at the maximum feasible minimum distance per length (d = 5, 8, 10, and 12, respectively; n = 96). Longer barcodes provide progressively higher accuracy; 30 bp barcodes reach ~97% at 95% identity. (B) Effect of distance metric during barcode generation: Levenshtein-generated tags (solid) outperform Hamming-generated tags (dashed) at equal nominal minimum distance, with a gap of 3–8 percentage points at 85–90% identity, owing to the higher realised inter-tag edit distance achieved by greedy selection in Levenshtein space (n = 48, end-mode, d = 8 for l = 20 bp and d = 12 for l = 30 bp). (C) Impact of full-mode position masks on demultiplexing accuracy for libraries with a 5′-end barcode (n = 96, l = 30 bp, d = 8, Levenshtein). End mode (reference) and full-mode with 5′ location information achieve very similar results, while full mode without a mask drops about 20% of accuracy. (D) Impact of full-mode position masks on demultiplexing accuracy for libraries with a mid-read barcode (n = 96, l = 30 bp, d = 8, Levenshtein). End mode (reference) achieves near-zero accuracy as expected, confirming correct mode-restriction behaviour. Full-mode search with a fractional mask (0.05:0.20) reaches ~45% accuracy at 95% identity; a fixed 5′ mask (5p:60) and full mode without a mask show intermediate and lower performance, respectively. (E) Scalability: accuracy as a function of barcode set size from n = 48 to n = 384, for dual-ended (circles) and spatial (diamonds) library types at three read identity levels (l = 30 bp, d = 8, Levenshtein). Accuracy varies by less than 2 percentage points across the full range, confirming applicability to high-throughput spatial transcriptomics and population-scale experiments.
At equal nominal minimum distance (d = 8) and tag length (20 bp), tags generated with Levenshtein distance constraints consistently outperformed Hamming-generated tags across all read identity levels, with a gap that widened at lower identity (+3–8 percentage points at 85–90% identity) ( Figure 4B). This advantage stems from the greedy selection algorithm achieving a higher actual minimum pairwise Levenshtein distance when operating in Levenshtein space: for d = 8 Levenshtein generation, the realised minimum inter-tag edit distance typically reached 12–15 bp, whereas Hamming-designed tags of the same nominal d achieved lower Levenshtein separations due to the Hamming–Levenshtein inequality. We therefore recommend Levenshtein generation for all ONT applications. Hamming-distance results are provided for comparison. At 30 bp and d = 12, both metrics converged at high identity (>95%), but Levenshtein remained superior at the clinically relevant 85–90% range.
For libraries where the barcode is positioned at the 5′ end of the read (spatial transcriptomics configuration, Figure 4C), end-mode demultiplexing was most effective (96.0% at 95% identity), as the tag is at the read terminus and is read with the lowest per-base error rate. Full-mode search with a 5′ position mask (5p:90, restricting the search window to the first 90 bp) matched end-mode performance (97.0% at 95% identity) and provided robustness to reads where the adapter truncated the tag start. Searching the full read without a mask reduced accuracy by 6–8 percentage points due to spurious off-target matches within the mRNA body, with a concomitant rise in misassignment to ~0.8% at 85% identity. This underscores the importance of correctly specifying the position mask to match the barcode location in the read.
For mid-read barcodes (DRS configuration, Figure 4D), end-mode correctly scored near-zero accuracy (~0%) as expected, confirming that the mode restriction works correctly. Full-mode with a 5p:90 mask achieved 98.0% accuracy at 95% identity. The fractional mask (0.05:0.20, covering 5–20% of read length) provided equivalent performance and is more portable across variable-length reads.
Despite increased difficulty of choosing from larger tag sets, accuracy was stable from n = 48 to n = 384 barcodes across all identity levels for both dual-ended and spatial library types, with less than 2 percentage points variation ( Figure 4E). At 95% identity, dual-ended end-mode accuracy remained above 97% up to 384 barcodes, and spatial full-mode (5p:90 mask) reached 97% at the same scale. At 85% identity, accuracy declined slightly with increasing n (from ~80% at n = 48 to ~76% at n = 384 for dual-ended), consistent with a greater probability of ambiguous assignment when the tag set is denser. These results confirm that taggen-demux scales to high-throughput spatial transcriptomics or population scale sequencing experiments without sacrificing accuracy.
The autoMaxDist parameter controls the maximum edit distance at which a read is accepted as matching a tag. The default adaptive rule (tag_length/5 for <20 bp, tag_length/4 for 20–29 bp, tag_length/3 for ≥30 bp) maximises read assignment while keeping misassignment below 0.5% for all recommended configurations (end-mode and full-mode with position mask) at read identities ≥85%. Users who require near-zero misassignment – for example in clinical or single-cell applications where sample cross-contamination must be minimised – should override the default with a more conservative threshold using the –max-dist flag. For instance, taggen-demux –max-dist 4 with 30 bp Levenshtein tags (d = 8) reduces the misassignment rate to <0.001% at the cost of leaving approximately 10–15% more reads unassigned at 85% identity (Extended Data Figure 4).
The following use cases demonstrate TagGen’s application to common long-read sequencing scenarios. Example input parameters are summarized in Table 5.
Input settings for barcode generation and demultiplexing across three representative library scenarios: high-error-tolerance direct RNA sequencing (UC1), older-chemistry ONT multiplexing (UC2), and single-cell spatial transcriptomics (UC3).
Introductory example: Standard ONT Sample Multiplexing
Scenario: A researcher needs to multiplex 96 samples for Oxford Nanopore sequencing using the standard flow cell configuration.
Input Parameters:
Barcode length: 14 bp
Target count: 96
Minimum Hamming distance: 4
GC content: 40–60%
Homopolymer limit: 3
CLI Command: taggen -n 96 -l 14 -d 4 --minGc 40 --maxGc 60 -r 3 -o ont_barcodes
Example Output (first 4 barcodes in ont_barcodes.fasta):
>Tag_001
ACGTACGTACGTAC
>Tag_002
TGCATGCATGCATG
>Tag_003
GATCGATCGATCGA
>Tag_004
CTAGCTAGCTAGCT
Generation time: 54 ms
Demultiplexing command: taggen --demux --tags ont_barcodes.fasta --reads reads.fastq --mode end --trim-mode ends
Expected performance: >97% correct barcode assignment at typical ONT error rates (10–15%).
Use Case 1: High-Error-Tolerance Direct RNA Sequencing
End-mode demultiplexing with custom adapter exclusion
Scenario: Direct RNA sequencing of degraded clinical samples where higher error rates are expected. Maximum error tolerance is required. 48-sample multiplexing experiment using a custom ligation adapter containing an 18 bp recognition sequence.
A common challenge in custom ONT library protocols is that proprietary or in-house adapters introduce short sequences that can cross-hybridise with naively generated barcodes, leading to spurious demultiplexing assignments. TagGen’s –exclude flag accepts a FASTA file of sequences to avoid; the greedy selection algorithm discards any candidate barcode whose edit distance to any excluded sequence falls below the minimum distance threshold, ensuring that the final tag set is orthogonal to the adapter.
For this scenario we recommend 24 bp Levenshtein tags with a minimum pairwise distance of d=10 to provide sufficient error tolerance. At 95% read identity – achievable with current R10.4.1/Kit14 chemistry – end-mode demultiplexing reaches 93.3% accuracy with near-zero misassignment. At 90% identity, accuracy is 83.5%, still suitable for standard discovery scale experiments.
Input Parameters:
Barcode length: 24 bp
Target count: 48
Minimum Levenshtein distance: 10
GC content: 45–55% (tighter for RNA stability)
Homopolymer limit: 2 (stricter for basecalling accuracy)
CLI Command: taggen -n 48 -l 24 -d 10 --metric levenshtein --minGc 45 --–maxGc 55 -r 2 --excludeFile custom_adapter.fasta -o uc1_tags -v
taggen --demux --tags uc1_tags.fasta --reads reads.fastq --mode end --trim-mode all
Generation time: ~30 ms
Expected performance: >98% correct assignment even at 20% error rate.
Use Case 2: Standard ONT Multiplexing with Older Chemistry
Scenario: A user has older-chemistry flow cells and needs to generate 96 barcodes that can still be confidently demultiplexed at high error rates.
Older R9.4.1 flow cells and high-molecular-weight or degraded samples routinely produce reads at 85–90% per-base identity. Under these conditions, strict end-mode demultiplexing is sensitive to read-start quality: partial exonuclease activity or pore clogging can introduce a few extra bases before the barcode, shifting it slightly from position 0. Full-mode search with a 5′ position mask (-position-mask 5p:60) restricts the alignment window to the first 60 bp of each read, tolerating this positional jitter while avoiding spurious matches in the downstream read body.
We recommend 30 bp Levenshtein barcodes (d=8) for this scenario. Benchmarks with simulated 5′-tagged reads show that full-mode with a 5p:60 mask achieves 84% accuracy at 85% read identity and 97% at 95% identity, matching or slightly exceeding end mode across the full identity range and maintaining misassignment below 0.1% in all conditions.
Input Parameters:
Barcode length: 30 bp
Target count: 96
Minimum Levenshtein distance: 8
Exclude sequences: Custom FASTA file containing adapter sequences
Exclude File (adapters.fasta):
>adapter_forward
AGATCGGAAGAGCACACGTCT
>adapter_reverse
AGATCGGAAGAGCGTCGTGTA
>custom_primer
TCGTCGGCAGCGTCAGATGTG
CLI Command: taggen -n 96 -l 30 -d 8 --metric levenshtein -f custom_adapter.fasta -o uc2_tags -v
taggen --demux --tags uc2_tags.fasta --reads reads.fastq --mode full --position-mask 5p:60 --trim-mode all
Generation time: ~95 ms
Expected performance: At 95% identity, full-mode with 5p:60 mask achieves 97% accuracy with near-zero misassignment; at 85% identity, accuracy is 84%, suitable for older chemistry experiments.
Use Case 3: Single-Cell RNA-seq with Spatial Barcodes (Zero Misassignment)
Scenario: Designing location barcodes for a custom spatial transcriptomics array with 384 capture spots, requiring error correction capability for imaging-based readout combined with nanopore sequencing.
In single-cell RNA-seq experiments where each barcode labels an individual patient sample or cell population, even a fraction of a percent of cross-sample contamination confounds downstream differential expression analysis. Here the barcode is embedded within a synthetic RNA spike-in added to each sample: during sequencing the tag appears mid-read, flanked by transcript sequence on both sides, at a position corresponding to roughly 5–20% of read length.
The fractional position mask (--position-mask 0.05:0.20) directs the search to this window regardless of read length, making the parameter portable across cells with varying transcript sizes. To achieve near-zero misassignment, we override the default acceptance threshold with --max-dist 4: at 90% read identity, this reduces misassignment from <0.1% (default) to <0.001%, at the cost of leaving approximately 10–15% more reads unassigned – a worthwhile trade when sample integrity is paramount.
Input Parameters: Barcode length: 30 bp
Target count: 384
Minimum Levenshtein distance: 8
GC content: 40–60%
Homopolymer limit: 3
CLI Command: taggen -n 384 -l 30 -d 8 --metric levenshtein --heatmap -o uc3_tags -v
taggen --demux --tags uc3_tags.fasta --reads reads.fastq \
--mode full --position-mask 0.05:0.20 \
--max-dist 4 --trim-mode all
Generation time: ~1.4 s. Expected performance: With –max-dist 4, misassignment is reduced to <0.001% at 90% read identity; approximately 10–15% of reads are left unassigned, which is acceptable when sample integrity is paramount.
The adoption of nanopore sequencing for transcriptomics, metagenomics, and clinical applications has outpaced the development of supporting bioinformatic tools. While barcode generators designed for Illumina sequencing work well at 8–12 bp, they cannot scale to the lengths required for error-tolerant nanopore barcoding.
TagGen (see Figure 5 for the graphical and command line user interface snapshots) addresses this gap through algorithmic innovation. By replacing O(4n) enumeration with O(k) sampling, we transform an exponentially scaling problem into a constant-time operation. The practical consequence is that TagGen succeeds – in milliseconds – at lengths where existing tools fail entirely. TagGen was designed to address a specific and underserved scenario in long-read sequencing: multiplexing experiments in which barcodes are not flanked by known primer or adapter sequences. In standard short-read or kit-based ONT workflows, demultiplexers such as minibar20 or Dorado21 leverage flanking adapter sequences as positional anchors, first locating the adapter and then extracting the adjacent barcode for comparison. This strategy works well when the library structure is fixed and the adapter sequence is intact. It fails – or cannot be applied at all – in custom library preparations, direct RNA sequencing, spatial transcriptomics capture plates, and synthetic RNA spike-in approaches, where either no consistent flanking sequence exists or the barcode can appear at variable positions within the read.
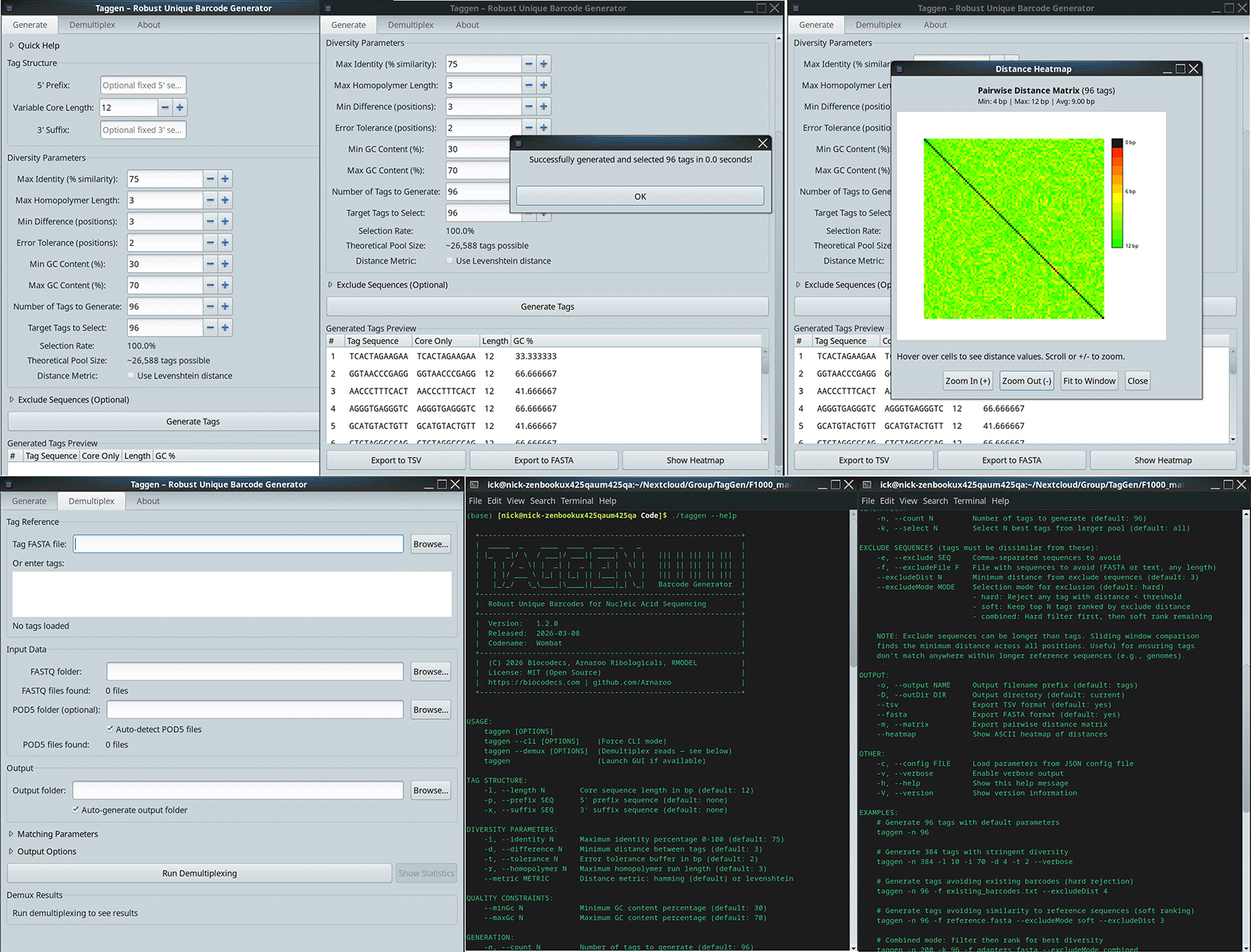
The composite screenshot shows four views of the TagGen application. Top left: the Generate tab, where users configure barcode parameters (tag length, count, minimum distance, GC content bounds, homopolymer limit, and exclude sequences) and preview generated barcodes with their core sequences, lengths, and GC content. Top right: the interactive pairwise distance heatmap displaying minimum Hamming or Levenshtein distances between all generated barcodes, enabling visual quality verification; generated barcodes can be exported in FASTA or TSV format. Bottom left: the Demultiplex tab, where users load barcode and FASTQ files, select search mode (end or full), configure position masks and acceptance thresholds, and view demultiplexing results. Centre: the command-line interface showing equivalent generation and demultiplexing commands with their options, demonstrating the full CLI workflow.
Our benchmarks confirm both sides of this distinction. In a head-to-head comparison on primer-flanked dual-ended reads – the use case for which minibar was designed – minibar achieves higher assignment rates than taggen-demux in anchor-free mode, particularly at longer barcodes and higher read identities (88–100% vs 59–97% at 80–95% identity, 30 bp barcodes). This is expected: primer anchoring is a genuinely more powerful strategy when the primer is present and intact. The comparison therefore does not identify a weakness in TagGen; it demarcates the two tools’ intended operating regimes. Where TagGen adds unique value is the large class of applications in which a primer anchor is absent, unreliable, or structurally variable – precisely the conditions under which minibar and analogous tools cannot be used.
Our systematic benchmarking across 154 simulated conditions (Panels A–E, Figure 4) provides the first comprehensive characterisation of anchor-free barcode demultiplexing performance as a function of tag length, minimum distance, error metric, position mask, and barcode set size.
The choice of error metric during tag generation has a direct and substantial effect on demultiplexing accuracy. Tags generated with Levenshtein distance constraints outperform Hamming-generated tags at equal nominal minimum distance, with a gap of 3–8 percentage points at 85–90% read identity. The mechanistic explanation is that the greedy selection algorithm, when operating in Levenshtein space, achieves a higher realised minimum pairwise edit distance between tags than the required minimum (typically 12–15 bp achieved vs 8 bp required for l = 30 bp, n = 96). Tags generated with Hamming d = 8 achieve lower realized Levenshtein separation due to the Hamming–Levenshtein inequality, leaving them more vulnerable to insertion and deletion errors characteristic of nanopore sequencing. We therefore recommend Levenshtein generation for all ONT applications. For standard 5′-tagged libraries, end-mode demultiplexing of 30 bp Levenshtein barcodes (d = 8, n = 96) achieves 96% accuracy at 95% read identity and 78% at 85% identity. Full-mode search with a 5′ position mask (−-position-mask 5p:60) matches or slightly exceeds end-mode performance and additionally tolerates reads in which the barcode is shifted from position 0 due to partial adapter degradation – a common occurrence with older R9.4.1 flow cells or degraded samples. This mode is recommended for high sample-count experiments on older chemistry where read starts are less uniform.
For mid-read barcodes – including tags embedded in synthetic RNA spike-ins for single-cell applications or tags placed internal to amplicons – end-mode demultiplexing correctly assigns near-zero reads, confirming that the mode restriction functions as a safety mechanism: reads are rejected rather than misassigned when the search window does not encompass the barcode position. Full-mode with a fractional position mask (−-position-mask 0.05:0.20) achieves 98% accuracy at 90% identity and is robust across variable read lengths. For zero-misassignment applications such as patient sample tracking in single-cell clinical studies, the –max-dist override reduces misassignment to <0.001% at a cost of approximately 10–15% more unassigned reads. Noise simulation provides the first systematic validation of barcode resilience under realistic nanopore error profiles. The results have direct implications for experimental design. Standard ONT sequencing (10–15% error): Barcodes ≥14 bp with d ≥ 4 achieve >97% resolution. Challenging conditions (20% error): Barcodes ≥20 bp with d ≥ 6 maintain >98% resolution. Direct RNA sequencing with degraded samples: 30 bp barcodes with d ≥ 8 provide near-perfect resolution even at 25% error. These findings suggest that researchers using nanopore sequencing should consider migrating from traditional 10–12 bp barcodes to 14–20 bp barcodes for routine applications, and to 24–30 bp barcodes when sample quality is uncertain.
TagGen’s capabilities enable several applications previously impractical due to computational limitations:
• Direct RNA sequencing: Full-length transcript barcoding with resilience to RNA degradation; anchor-free demultiplexing locates mid-read barcodes without adapter sequences
• Spatial transcriptomics on ONT: Location barcodes robust to platform-specific errors, with fractional position masks enabling demultiplexing across variable-length reads
• Environmental metagenomics: Sample multiplexing for long-read community profiling
• Clinical nanopore diagnostics: High-confidence sample identification in point-of-care settings, with configurable acceptance thresholds to achieve near-zero misassignment rates
Several limitations should be considered when using TagGen.
Distance metric: TagGen supports both Hamming and Levenshtein distance for barcode generation, and uses Levenshtein distance for demultiplexing. While the validation results confirm high accuracy under indel-rich nanopore error profiles, future work could explore probability divergence15 as an alternative metric for selection, which may offer additional gains in barcode separability for high-error profiles.
Optimality: The greedy selection algorithm produces locally optimal but not globally optimal barcode sets. For most practical applications with 96–384 barcodes, this approximation is sufficient, but users requiring mathematically optimal sets for very large barcode counts may need to employ more computationally intensive methods such as integer linear programming.
Memory scaling: While TagGen avoids the exponential memory requirements of exhaustive enumeration, very large candidate pools (>1 million sequences) for extremely long barcodes may still require substantial RAM. The default candidate pool size is tuned for typical applications.
TagGen represents the first barcode generator capable of producing error-tolerant barcodes for long-read sequencing applications. Key contributions include:
1. Algorithmic advance: Monte Carlo sampling replaces exponential enumeration, enabling barcode generation at 14–30 bp
2. Validated resilience: Systematic demonstration that longer barcodes maintain resolution under nanopore error profiles
3. Practical utility: Sub-second generation times for lengths where existing tools fail
4. Anchor-free demultiplexing: An integrated demultiplexer that locates barcodes at any read position without requiring adapter sequences, validated across 154 parameter combinations with near-zero misassignment.
5. Dual interface: Both GUI for interactive use and comprehensive CLI for automated pipelines
As nanopore sequencing expands into transcriptomics, diagnostics, and field applications, TagGen provides researchers with the tools to design robust, validated barcodes matched to their platform’s error characteristics and library architecture.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)