Keywords
Multiple myeloma, dormancy, 3D culture, polyHIPE scaffold, bone microenvironment, osteoblasts, 3Rs, drug response.
This article is included in the NC3Rs gateway.
Multiple myeloma, dormancy, 3D culture, polyHIPE scaffold, bone microenvironment, osteoblasts, 3Rs, drug response.
Multiple myeloma is a plasma cell malignancy within the bone marrow (BM).1,2 It is the third most common haematological cancer in the UK, and many current therapies, such as chemotherapy, immunotherapy, and targeted treatments, have helped make significant strides in treating the disease.3,4 However, a significant challenge in myeloma treatment is disease relapse, with approximately 66% of myeloma patients relapsing within four years of first remission.5 A major clinical challenge in relapse prevention is the persistence of chemotherapy-resistant cells, including dormant myeloma cells (DMCs), during minimal residual disease (MRD).6–8 DMCs are non-proliferative cells that are resistant to conventional treatments and reside in specialised endosteal niches within the BM, making them difficult to target.6,7 Much of what is known about DMC biology comes from preclinical studies, both in vitro and in vivo, 9,10 where it has been demonstrated that their dormant status can be controlled and influenced by interactions with niche-resident cells, including osteoblast-lineage cells and osteoclasts.6,7,10 Consequently, DMCs may escape growth arrest and repopulate the BM, leading to disease relapse. Therefore, developing therapies that specifically target DMCs is critical for improving the long-term survival outcomes of patients with myeloma.
Several models have been employed to study dormant cancer cells, including in vitro and in vivo systems; these have been used to further our understanding of dormancy mechanisms and to investigate drug effects. One of the best-characterised murine models of myeloma is 5TGM1, where murine myeloma 5TGM1 cells are injected via the tail vein into C57BL/KaLwRijHsd mice and home to the BM.11–14 This model has been used to demonstrate that DMCs respond differently to chemotherapy agents, exhibiting resistance to melphalan and bortezomib.6 Additionally, AXL inhibitors such as cabozantinib have been shown to release DMCs from endosteal niches in this model.7
While in vivo models have been fundamental for testing the effects of therapeutic agents on DMCs and their endosteal niche interactions, they have significant limitations. They require a large number of animals to be used, which raises ethical concerns and limits drug testing capabilities owing to time and cost constraints. Additionally, the severity of these models is often defined as moderate under the Animals Scientific Procedures Act 1986 because of the tumour burden placed on the animals. Moreover, in vivo models often use only a single cell line for tumour inoculation, limiting their ability to capture the high heterogeneity of myeloma. Therefore, this places a large limitation on the interpretation of data generated from animal studies.
In contrast, in vitro models offer more ethical and cost-effective alternatives. They have been used to understand the molecular mechanisms driving dormancy. For instance, in vitro, DMCs can be observed in monocultures with low prevalence, but their prevalence can be increased by co-cultures such as osteoblasts through both direct cell-to-cell contact and indirect signalling and reduced through indirect effects of mesenchymal stem cells.6,7,10,15 Established myeloma cell lines as well as primary myeloma cells from patients can be used to identify key markers of dormancy. In vitro models can overcome some limitations of in vivo models by incorporating a more heterogeneous sample pool and better replicating the complexity of the BM microenvironment. However, drug response data collected from two-dimensional (2D) in vitro models often fail to match the clinical outcomes.16,17 Furthermore, although technically simpler, 2D in vitro systems fail to recreate a three-dimensional (3D) bone microenvironment, which is crucial for in vivo myeloma cell dormancy. This limitation in the models can lead to contrasting data when drug candidates are considered, thereby slowing treatment development.
To address these challenges, we developed a novel in vitro model that mimics in vivo osteoblast and myeloma cell interactions in a 3D environment. This model provides a platform for initial drug screening against DMCs and mechanistic studies prior to the use of more focused preclinical in vivo studies, thereby significantly reducing the total number of animals used in myeloma research. This is because, on average, 50 mice were used in each myeloma in vivo study, and ~ 140 papers were published annually. By providing a controllable in vitro system for early drug response studies, this model has the potential to replace small pilot in vivo studies typically involving 10–20 mice per study, which are often used before larger efficacy in vivo studies.
Our model utilises polycaprolactone (PCL), which is commonly used in tissue engineering applications, to provide a 3D scaffold foundation that offers structural support similar to that of in vivo bone.18,19 PCL is a synthetic biodegradable polymer that has previously been validated extensively in human bone regeneration, making it a favourable material that is cost-effective and readily available.20,21 A key advantage of using PCL is that it eliminates the need to use animal-derived products, unlike similar 3D bone models such as those built on Matrigel, which can be unreliable owing to batch-to-batch variability.
While in vitro 3D models have been used to study myeloma, none have used PCL or specifically assessed DMCs due to various limitations. For example, most 3D models rely heavily on imaging techniques to examine cells within the model because of the limited retrieval of cells. The use of flow cytometry analysis is often restricted for 3D models because of the necessary steps required to isolate cells that can alter cellular characteristics.
By manufacturing the scaffold, the base material is processed to produce a PCL-based polymerised High Internal Phase Emulsion (polyHIPE) scaffold structure.18,20,22 This provided a highly porous, interconnected internal architecture that facilitated cell migration and simulated the BM niche. The polyHIPE structure can also be highly attuned to generate a refined scaffold that supports cell growth and cellular dormancy. Importantly, cells can be isolated from scaffolds through enzymatic digestion steps similar to those used in standard 2D culture methods.
To make our model easily achievable and adaptable, few additional components were added to the scaffold base. First, osteoblast-lineage cells can be cultured directly on the scaffold material to facilitate their ingrowth throughout the pores to mimic the endosteal niche. Following this, myeloma cells can be incorporated onto the scaffold, and their proliferation is monitored through the retention of a cell membrane dye, extensively used by others as a method to track dormant cells.6,7 As a result, our model offers a more robust and customisable system to replicate key aspects of the in vivo endosteal niche, offering potential insights into dormancy mechanisms.
In this article, we provide an overview of how we have developed and optimised this model, how the model (henceforth termed polyHIPE scaffold) can be prepared for in vitro systems, and how it can be applied to answer various research questions. We will also provide a step-by-step protocol covering the manufacturing of the PCL base, culturing osteoblast-lineage cells and myeloma cells on the scaffold, and tracking DMCs through several experimental procedures.
We aimed for this model to be used by myeloma researchers focused on drug testing against DMCs, but it could also be applied more broadly as a versatile 3D model to study the biology and therapeutic responses of myeloma and niche cells. Moreover, it could be applied more generally to researchers investigating tumour–bone interactions, stromal niche biology, or early-stage therapeutic responses within 3D microenvironments. We propose that this model offers an easier alternative to gel-based models owing to its simplicity and reproducibility.
The supplier and reference ID of all the reagents and equipment used in this study are listed in Table 1. The methods described here focus on a small selection of myeloma and osteoblastic cells, but the model can be readily adapted to accommodate other cell lines/types.
| Chemicals and reagents | ||
|---|---|---|
| Reagent | Source | Identifier* |
| RPMI 1640 Medium, GlutaMAX™ Supplement | GIBCO, Life Technologies | Cat#61870–010 |
| Fetal Bovine Serum (FBS) | ThermoFisher | Cat#10500064 (Lot: 2575614H) |
| MEM Non-Essential Amino Acids Solution | Fisher Scientific | Cat#25–025-CIR |
| Sodium Pyruvate | ThermoFisher | Cat#11360–070 |
| Penicillin-Streptomycin (pen/strep) | ThermoFisher | Cat#15140–122 |
| DMEM/F-12, GlutaMAX™ | ThermoFisher | Cat#10565018 |
| Geneticin™ Selective Antibiotic (G418) | ThermoFisher | Cat#10131035 |
| Oxoid™ Phosphate Buffered Saline Tablets (PBS) | ThermoFisher | Cat#BR0014G |
| Vybrant™ DiD Cell-Labeling Solution (DiD) | ThermoFisher | Cat#V22887 |
| AlamarBlue™ Cell Viability Reagent | ThermoFisher | Cat#DAL1100 |
| Paraformaldehyde, 4% in PBS | ThermoFisher | Cat#J61899 |
| LIVE/DEAD™ Fixable Violet Dead Cell Stain Kit | ThermoFisher | Cat#L34955 |
| DAPI Solution | ThermoFisher | Cat#62248 (Lot: XD3562132) |
| Bright Cryo-M-Bed (OCT) | Bright Instruments | Cat#53581 |
| Andwin Scientific Cryomould | Fisher Scientific | Cat#NC9511236 |
| Epredia™ SuperFrost Plus™ Adhesion Slides | Fisher Scientific | Cat#12312148 |
| Accutase® Cell Detachment Solution | Innovative Cell Technologies | Cat#AT104 |
| Trypsin-EDTA | ThermoFisher | Cat#25300062 |
| Hypermer B246 (surfactant) | Croda | https://www.crodaindustrialspecialties.com/en-gb/product-finder/product/531-hypermer_1_b246 |
| PCLMA | In house | https://licensing.sheffield.ac.uk/product/polycaprolactone-methacrylate-pclma |
| 2,4,6-Trimethylbenzoyl Phosphine Oxide/2-Hydroxy-2- Methylpropiophenone (Photoinitiator) | Sigma Aldrich | Cat#405663 |
| Chloroform | Sigma Aldrich | Cat#02487 |
| Toluene | Sigma Aldrich | Cat#89680 |
| Experimental models: polyHIPE | ||
|---|---|---|
| Item | Source | Identifier |
| PCL polyHIPE discs | TUoS | N/A |
| Software | ||
|---|---|---|
| Software | Source | Identifier/Link |
| FlowJo v10 | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo
RRID:SCR_008520 |
| GraphPad Prism v9.0 | GraphPad | https://www.graphpad.com/
RRID:SCR_002798 |
| ImageJ Fiji | NIH | https://imagej.nih.gov/
RRID:SCR_003070 |
PolyHIPE scaffolds were prepared in-house as previously described.20,22 Briefly, surfactant was dissolved by heating in a mixture of 4-arm polycaprolactone methacrylate, then cooled prior to the addition of a photoinitiator and a solvent blend (40% toluene, 60% chloroform). Full details of the quantities used for a single batch are provided in Protocol 1 below; however, these can be scaled proportionally as required by users. Using a magnetic stirrer, the mixture was mixed at 400 rpm for 3 min at 37 °C, and then 2 ml water was added dropwise over 3 min. The resulting emulsion was mixed for an additional 5 min and then cured in a syringe for 5 min on both sides to produce polyHIPE tubes. This was then washed in 100% methanol for three days, then in water for a further day in order to remove contaminants. The washed polyHIPEs were then dried in a vacuum oven at room temperature overnight prior to sectioning to the desired thickness using a vibratome and then maintained in a dry environment at room temperature until required (Fig. 1). We observed that a thickness of 250 μm was ideal; however, this thickness could be tailored by users.
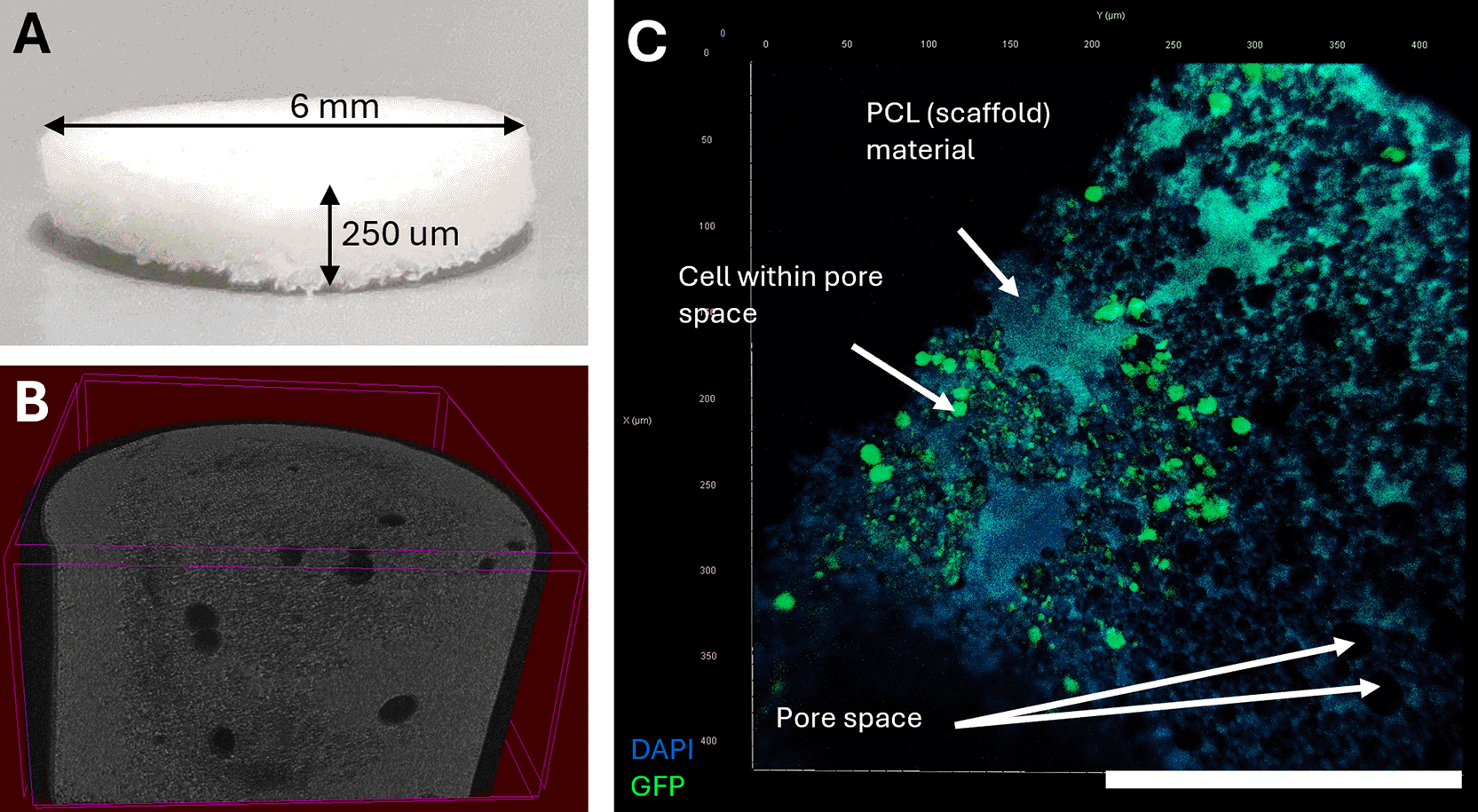
(A) Photograph of a PolyHIPE scaffold with sponge-like appearance post processing cut to shape, with parameters of 6 mm diameter and 250 μm thickness. The highly porous structure and interconnectivity can be observed by (B) CT imaging to generate a whole 3D structure and (C) fluorescent imaging to capture a higher magnification cross section of a scaffold.
Three myeloma cell lines (U266, JJN3, and 5TGM1) and three osteoblastic cell lines (MG-63, hFOB 1.19, and MC3T3) were used ( Table 2). Myeloma cell lines JJN-3 (DSMZ, Germany), U266 (LGC Standards, UK), and 5TGM111 were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin (P/S), 1% non-essential amino acids (NEAA; Corning, UK), and 1% sodium pyruvate (SP).
| Cell name* | Species | Cell type | Description |
|---|---|---|---|
| U266-GFP-Luc | Human | Myeloma | Stably transduced to express luciferase (luc)-2A-green fluorescent protein (GFP). Doubling time ~ 50-60 h. Derived from peripheral blood of a 53-year-old male with refractory multiple myeloma. (RRID:CVCL_0566) |
| JJN3-GFP-Luc | Human | Myeloma | Cells stably express luciferase (luc)-2A-green fluorescent protein (GFP). Doubling time ~ 20-35 h. Derived from primary site tumour within the bone marrow of a female 57-year-old plasma cell leukeamia patient. (RRID:CVCL_2078) |
| 5TGM1-GFP-Luc | Mouse | Myeloma | Cells stably express luciferase (luc)-2A-green fluorescent protein (GFP). Derived from murine 5T33 cells after repeated cycles of injection and harvesting from the bone marrow of C57BL/KaLwRij mice. (RRID:CVCL_VI66) |
| hFOB 1.19-RFP-Luc | Human | Foetal osteoblasts | Human foetal osteoblasts derived from foetal bone. Doubling time ~ 36 h. (RRID:CVCL_3708) |
| MG-63 | Human | Osteosarcoma | Human osteosarcoma cell line derived from a14-year-old male. Fibroblastic morphology described to have an immature osteoblast-like phenotype. (RRID: CVCL_0426) |
| MC3T3 | Mouse | Pre-osteoblast | Murine pre-osteoblast cell line with the capability of differentiating into osteoblasts and osteocytes. Described to have a mature osteoblast-like phenotype. (RRID: CVCL_0D74) |
MG-63 (ATCC) and MC3T3-E1 subclone 4 (ATCC) cells were maintained in minimum essential medium (MEM) alpha supplemented with 10% FBS and 1% P/S. Human fetal osteoblasts (hFOB 1.19; ATCC) were maintained in Dulbecco’s Modified Eagle’s medium/nutrient mixture F-12 medium supplemented with 10% FBS, 1% P/S, and 0.3 mg/ml G418 sulphate.
All cells were maintained at 37 °C with 5% CO2 in a humidified incubator (95 ± 5% relative humidity), except hFOB 1.19 monocultured cells, which were maintained at 34 °C. The co-cultures were maintained in complete RPMI medium for myeloma cell maintenance.
All cells were routinely tested for mycoplasma contamination prior to use and were confirmed to be mycoplasma-free. Human cell lines were authenticated by short tandem repeat (STR) profiling using ATCC profiling services. Murine cell lines were obtained from established academic or commercial sources. Cells were expanded from early passage stocks and used within 30 passages, relative to the passage number provided by the supplier, to minimise phenotypic drift. Unless stated otherwise, cells were maintained under standard culture conditions and used during the logarithmic phase of growth.
Scaffolds were sterilised by immersion in 100% methanol for 24 h, followed by washing in either phosphate-buffered saline (PBS) or complete media for 24 h, with a minimum of three changes. The scaffolds were then transferred to 48-well plates, and excess liquid was removed. Osteoblast cells were seeded onto the surface of scaffolds in 20 μl of RPMI media, at a density of 50,000 cells, and then incubated for 2 h at 37 °C to allow cell attachment. Following incubation, media was added to the wells containing scaffolds, and the models were maintained as required. Osteoblasts were cultured for 3 days, then the scaffold was transferred to a new 48-well plate, and excess liquid was removed. Myeloma cells were subsequently seeded onto the polyHIPE scaffold surface in the same manner as osteoblasts (Fig. 2).
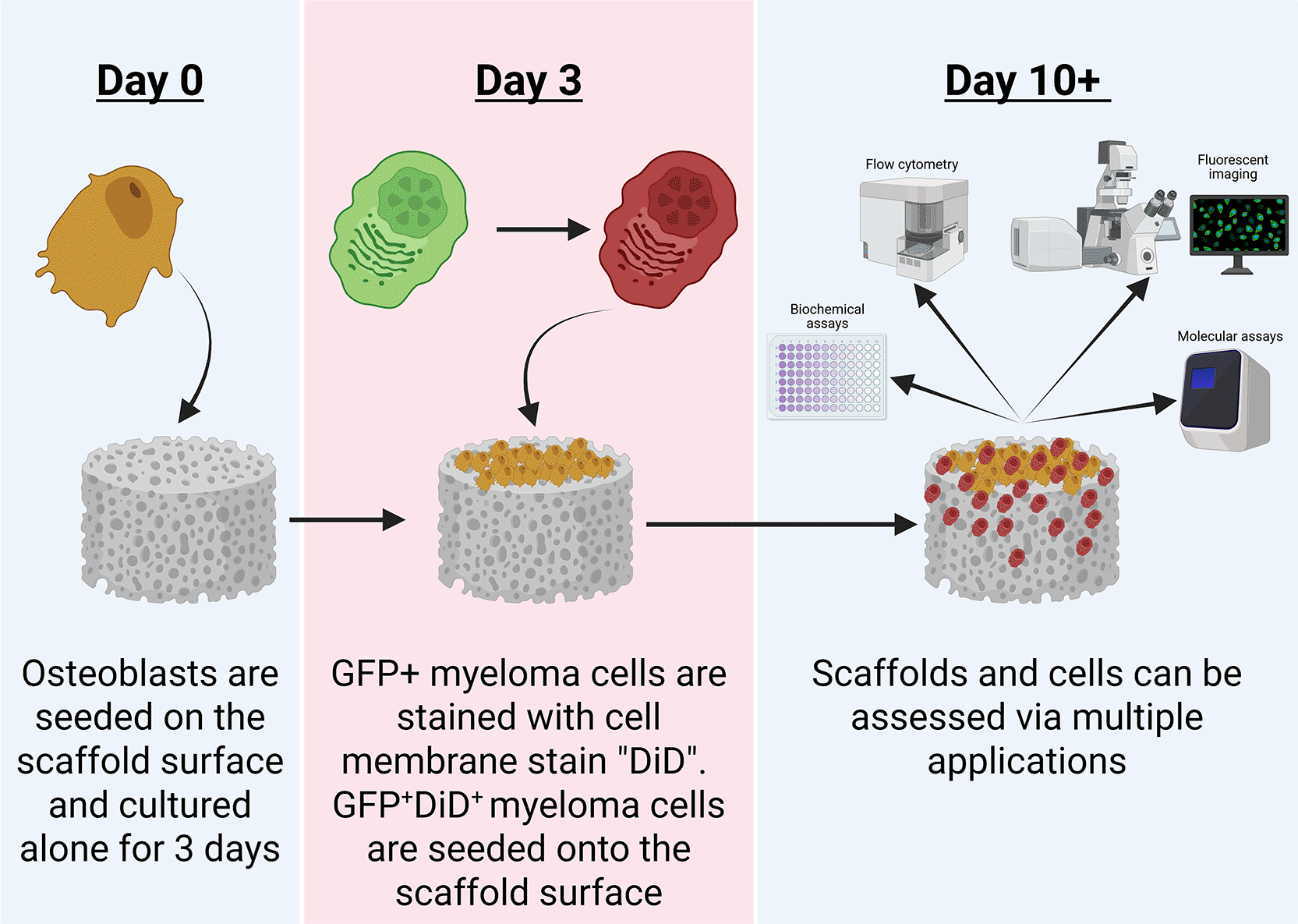
Osteoblasts are first applied to the scaffold, then allowed to adhere and migrate from the scaffold surface over 3 days of “pre-seeding”. Following this, myeloma cells are stained with a cell membrane label (DiD) and seeded onto the scaffolds. Scaffolds are the maintained up to day 10+ prior to further downstream assessments. Figure created in BioRender.24
Osteoblastic (MC3T3) and myeloma (5TGM1) cells were seeded onto scaffolds as described above and co-cultured for 21 days post-myeloma cell seeding. Scaffolds were then washed in room temperature PBS x3, fixed in 4% PFA for 15 min at room temperature, then washed in PBS x3. Since the scaffolds are auto-fluorescent when imaged using a 405 nm laser, to increase the fluorescent signal of the material, scaffolds were then stained with DAPI (1:1000) for 10 min at room temperature and washed in PBS x3. The scaffolds were then dipped in OCT and placed into a cryomold containing a layer of OCT. This was then submerged in liquid nitrogen for 15 s and quickly transferred to a steel chuck in a cryostat chamber set to −20 °C. Sections were cut to a thickness of 10–20 μm, with 10 μm representing the minimum thickness required to preserve structural integrity, and 20 μm the maximum thickness to ensure sufficient optical resolution and signal clarity for confocal microscopy. Sections were mounted on SuperFrost Plus glass slides at room temperature and air-dried for a minimum of 3 h.
Fluorescent imaging of the scaffolds was performed using a Zeiss LSM980 confocal microscope. Images were captured at x10 or x20 magnification using Plan-Apochromat 10x/0.3 M27 air or 20x/0.8 M27 air objectives, respectively. DAPI, GFP, and DiD channels were excited with 405, 488, and 639 nm lasers, respectively. The laser power, detector gain, and pinhole settings were optimised to maximise the signal while avoiding detector saturation and were kept consistent across samples. Images were acquired using Zeiss Zen 3.5 (blue edition) software.
Osteoblastic (MG-63 or hFOB) and myeloma (JJN3 or U266) cells were seeded onto scaffolds as described above, 24-hours post myeloma cell seeding a drug was added to the culture well and scaffolds cultured for a further 3 days. For JJN3 co-cultures, melphalan (5.8 μM), bortezomib (1.5 nM), or lenalidomide (5 μM) were used at concentrations representing 2D IC25 values. For U266 co-cultures, bortezomib (3 nM), lenalidomide (50 μM), or a combination of both were used at concentrations representing 2D IC50 values. The study design incorporated consistent seeding densities and repeated biological replicates to ensure reproducibility. Randomisation and blinding were not applied in this workflow, as all handling and measurements were performed using standardised protocols to maintain consistency across samples. Randomisation and blinding were implemented for scaffold-based flow cytometry assays, where individual scaffolds were processed and measured separately.
AlamarBlue® assessment
For JJN3 cultures, mixed-population cell viability was assessed using the alamarBlue® assay (Fig. 4A). 2D controls were maintained and treated in a similar manner in 6-well plates. Following the seeding of cells on scaffolds, at the indicated time points, scaffolds were moved to new wells containing fresh medium. The movement of scaffolds to new wells was necessary to ensure that measurements were reflective only of cells growing on and within the polyHIPE scaffolds, rather than those that failed to attach/fall off the scaffold. Once removed, 10% (v/v) alamarBlue® reagent was added to wells, and the scaffolds were incubated at 37 °C for 4 h. Following this, scaffolds were removed from the wells, and the fluorescence produced by the reduction of the alamarBlue® reagent in the remaining media was measured using an EnSight Multimode Plate Reader. Relative fluorescence intensity, commonly used as a proxy for cell viability in drug response assays, was used as an indicator of cell metabolic activity. Values were normalised to those of vehicle controls and expressed as percentages.
Flow cytometry assessment
For U266 co-cultures, cells were enzymatically removed from scaffolds following a regimen of 10 min wash in PBS, followed by 30 min incubation at room temperature on a shaker with accutase, and 10 min incubation with trypsin-EDTA at 37 °C on a shaker. PBS, accutase, and trypsin suspensions were collected after each step, and the cells were collected by centrifugation at 800xg for 5 min. As a further step to maximise the total number of cells isolated from scaffolds, the scaffolds were cut into segments and centrifuged in PBS at 800xg for 5 min.
Whole myeloma cell and DMC (DiDHi) cell population viability was assessed by flow cytometry (LSRII flow cytometer) following 30 min of incubation with live/dead violet at room temperature.
This study did not involve human participants or human tissue samples requiring ethics approval. All human cell lines used in this study were obtained from commercial or academic sources and were used in accordance with institutional guidelines.
Unless otherwise stated, all experiments were performed in which a single scaffold represented an experimental unit (n). Data were analysed using GraphPad Prism v9.0 (GraphPad Software, USA) and are expressed as the mean ± standard deviation. Pairwise comparisons were performed using two-tailed Student’s t-tests. Statistical significance was defined as P < 0.05.
Below is a step-by-step guide for developing, applying, and troubleshooting the polyHIPE model for dormancy assessment. It is broken down into five protocols with a sixth section focused on troubleshooting.
PolyHIPE scaffolds were synthesised according to the protocols of Aldemir Dikici et al.19 and Jackson et al., 20 by forming a water-in-oil high internal phase emulsion (HIPE), with a 40:60 aqueous-to-oil phase ratio to ensure high porosity, which is then photopolymerised to a polyHIPE. Solvents and uncured materials were extracted prior to sectioning the discs and sterilisation for cell culture.
Steps:
1. Prepare PCL-M emulsion (~ 30 minutes)
a. In a glass vial, 0.40 g of PCL-M and 0.04 g of surfactant were mixed together. The surfactant was gently heated and allowed to cool to room temperature.
b. Add 0.03 g Photoinitiator and 0.60 g of solvent blend (60 wt. % chloroform: 40 wt. % toluene) was added to the cooled mixture. Keep protected from light.
c. A magnetic stirrer (20 mm × 7 mm) was used to stir the mixture at 400 rpm and 37 °C for 3 min to ensure that a homogeneous oil phase was produced.
d. Add 2 ml of deionised water dropwise over 3 minutes while maintaining constant stirring.
e. Mix for an additional 5 minutes to stabilise the emulsion.
CRITICAL: Maintain temperature and stirring speed throughout to ensure reproducible pore architecture, lower stirring speeds will produce larger pores.
2. Polymerise the emulsion (~15 minutes)
a. Load the emulsion into a 2 ml syringe (of ~6 mm diameter).
b. Cure using an OmniCure Series 1000 system for 5 min on each side (18 W/cm2, spectral output of 250–600 nm).
c. Remove the polymerised polyHIPE tube from the syringe.
NOTE: We use 2 ml syringes to produce an ideal diameter for 48-well plates. Silicone moulds can also be used to cure the emulsion but may interfere with later processing steps when a glue is required.
3. Washing and drying (6 days)
a. Submerge polyHIPE tubes in 100% methanol for three days, replacing methanol every 24 h.
b. The mixture was washed in deionised water for 3 days, and the water was changed every 24 h to remove excess surfactant, solvent, and residual monomer.
c. The scaffolds were dried overnight in a vacuum oven at room temperature until they were fully solvent-free.
d. Scaffolds can be stored in this dry state long term (6 months).
4. Sectioning and sterilisation (2–3 days)
a. Using a scalpel blade, dried polyHIPE tubes (~1 cm tall) were cut into smaller trunks to facilitate easier cutting in the next step.
b. Using a small dot of quick drying glue, attach the polyHIPE tube standing on one of its circular ends to a vibratome steel chuck, and let the glue dry.
c. Using a vibratome (frequency of 80 Hz, amplitude of 1.00 mm and speed of 0.1–0.3 mm/s), section polyHIPE trunks into scaffold discs of 250 μm thickness.
d. Dip discs in deionised water or PBS and lay flat on a sealable container (e.g., petri dish) and allow to air-dry overnight. Failure to do this may result in curled discs.
e. Store dry until required for experiments.
f. For sterilisation, immerse discs in 100% methanol for 24 h (preferred) or 70% ethanol (alternative), followed by three PBS washes for a minimum of 1.5 h (ideally over 24 h to ensure complete removal of alcohol).
g. Store scaffolds submerged in sterile PBS or media at room temperature for no longer than 24 h or proceed directly for cell seeding.
NOTE: Do not press the polyHIPE trunk into the glue with too much force as this may result in glue absorbing into the polyHIPE.
NOTE: The first cut into a polyHIPE trunk should be used to level off the top of the sample so that a straight edge is produced, this first cut should therefore be discarded.
NOTE: The given quantities in this protocol are appropriate to generate 200–300 scaffolds of 250 μm.
NOTE: We used UHU 3–62686 Super Glue Ultra-Fast Liquid to attach polyHIPE tubes to steel chucks but believe any fast-acting glue would be sufficient.
Osteoblast cells (hFOB 1.19) were pre-seeded onto the scaffolds to generate the osteoblast component of the 3D model. Pre-seeding prior to myeloma cells facilitates sufficient attachment and migration into the scaffolds such that myeloma cells do not outcompete osteoblasts because of their shorter doubling times. Osteoblasts were cultured for three days prior to the addition of myeloma cells, but osteoblasts could be cultured for longer periods (we cultured osteoblasts alone for up to 28 days).
Steps:
1. Scaffold preparation (~15 minutes)
a. Scaffolds were removed from the wash medium 10–15 minutes prior to seeding hFOB 1.19 cells onto scaffolds, then placed into individual wells of a 48-well tissue culture plate.
b. Excess medium carried over with the scaffold was removed from the wells, and the scaffolds were placed so that the attachment of osteoblasts onto the scaffolds was not affected by excess liquid.
c. Scaffolds were maintained at room temperature or placed in an incubator at 37 °C until the cells were ready to be seeded. This facilitated better liquid retention when cells were added to the scaffold, aiding cell attachment.
2. Osteoblast cell preparation (~20 minutes)
a. Culture hFOB 1.19 cells at 34 °C until ~80% confluence in a T75 tissue culture flask.
b. Remove media and wash the cell surface with 10 ml PBS twice.
c. Add 1–2 ml trypsin-EDTA (0.25%) to the T75 flask and incubate at 34 °C for 5 min.
d. Add 8–9 ml of complete medium to quench the trypsin and resuspend the cells.
e. Count cells using a hemocytometer or automated cell counter with trypan blue exclusion to ensure high viability (>90%) before seeding onto scaffolds.
f. Centrifuge cells at 300xg for 5 min and resuspend at a concentration of 2.5x106 cells/ml such that 20 μl contains 50,000 cells.
3. Seeding onto scaffolds (2 hours)
a. Seed cells on top of the scaffolds by carefully pipetting a 20 μl droplet onto the centre of the surface of each scaffold to promote attachment to the scaffold rather than the plastic surface of the well.
b. In the spare wells surrounding the scaffolds, PBS was added to prevent the scaffolds from drying out, and the scaffolds were placed in an incubator set to 34 °C for 2 h to allow cells to attach to the scaffold.
NOTE: This seeding strategy is necessary for adherent cell lines, where a proportion of cells may otherwise adhere to the well plate surface. The high localised density of cells in a small volume is important to improve scaffold-specific seeding/attachment efficiency.
4. Post-seeding culture (3–28 days)
a. After the initial 2-hour incubation to facilitate hFOB 1.19 cell attachment, 500 μl of complete medium was gently added to the wells to fully submerge the scaffolds. Adding media along the inner edge of the wells minimises the disruption of cells on the scaffolds.
b. Scaffolds should be maintained at 34 °C with 5% CO2 for 72 h until required for subsequent use.
NOTE: Do not use 96 well plates for culture since the medium volume is not sufficient for the cell number seeded onto scaffolds. Similarly, scaffolds can be seeded into larger well plates but will float in the excess medium within these wells. Therefore, we used 48 well plates and replaced the media every 1–2 days as required.
NOTE: Here, scaffolds are maintained at 34 °C since this is the optimum maintenance temperature for hFOB 1.19 cells, if other osteoblast cells are used the temperature should be altered to match the cell’s required temperature.
Vybrant DiD cell labelling solution (DiD) is used to indirectly track cell proliferation, since non-proliferative cells retain a strong dye signal, whereas proliferating cells do not. Therefore, we used DiD to identify DMCs (e.g. JJN3), since these cells retain a strong DiD signal. We used DiD, but other similar dyes could also be used. The myeloma cells were labelled prior to seeding onto scaffolds; we did not observe DiD transfer onto the polyHIPE material, but it is possible for DiD to transfer to osteoblasts.
Steps:
1. Myeloma cell preparation (~15 minutes)
a. JJN3 cells were maintained at a density of 0.4-1x106 cells/ml at 37 °C in a T75 tissue culture flask and passaged upon reaching 1x106 cells/ml to maintain logarithmic growth.
b. Myeloma cells can be counted in suspension directly from their culture flask using a hemocytometer or automated cell counter with trypan blue exclusion to ensure high viability (>90%) before seeding onto scaffolds.
c. The required number of cells was centrifuged at 500xg for 5 min in a universal tube and the cell pellet was retained.
d. The cell pellet was resuspended in a volume of serum-free media at a concentration of 1x106 cells/mL.
NOTE: We recommend staining double the number of cells required since loss of cells is commonly observed during the following steps, and an excess of cells is required as a reference point marker of the highest DiD mean fluorescent intensity (MFI) for flow cytometric analyses.
NOTE: The next steps followed the manufacturer’s instructions but should be optimised for each cell line used.
2. DiD staining (1 hour)
a. DiD reagent was added to the cell suspension at a volume of 5 μl per 1 ml of serum-free medium (or 5 μl per 1x106 cells).
b. The cells were incubated with DiD at 37 °C for 20 min. The tube was gently shaken after the first 10 min to avoid cells settling at the bottom of the tube, resulting in nonuniform staining.
c. Following incubation, the cells were centrifuged at 600xg for 5 min and the supernatant was discarded to remove excess unbound DiD dye.
d. The cell pellet was washed in serum containing medium (1x106 cells/mL) and centrifuged for 5 minutes at 600xg.
e. Repeat step 8 a total of three times to ensure that all excess DiD is removed.
f. Following the last wash, the cells were resuspended in an appropriate volume of serum medium and counted.
g. The volume of media needed to suspend cells was calculated to achieve a concentration of 2.5x106 cells per ml (5x104 cells per 20 μl).
h. Seed DiD-labelled myeloma cells onto scaffolds as described below.
NOTE: We increased the centrifugation speed following DiD labelling as the dye reduced the pelleting of cells at lower speeds.
NOTE: It is not necessary to perform these steps in a universal tube, but the flat bottom facilitates standing in an incubator and centrifugation without the need to transfer the cells between containers.
Following osteoblast pre-seeding on the scaffold for three days, (DiD labelled) myeloma cells (JJN3) were added to the model system. We have cultured myeloma cells with osteoblasts for up to 21 days and advise leaving myeloma cells for at least 24 h for shorter study periods.
Steps:
1. Scaffold preparation (~10 minutes)
a. After 3 days of incubation with osteoblasts in monoculture, the scaffolds were transferred into clean wells of a fresh 48-well plate to avoid contamination from osteoblasts that had adhered to the original well plastic surface.
b. Scaffolds were moved 10 min before myeloma cells were seeded. Excess medium was removed from the wells, and scaffolds were placed back into an incubator at 37 °C until DiD-labelled myeloma cells were added.
2. Myeloma cell preparation and seeding onto scaffolds (2 hours)
a. DiD labelled myeloma cells should be at a concentration of 2.5x106 cells/ml such that 20 μl contains 5x104 cells (Protocol 3).
b. The same method used to seed osteoblasts was used; myeloma cells were placed on top of the scaffold by carefully pipetting a 20 μl droplet onto the centre of the scaffold surface to promote attachment to the scaffold rather than to the plastic surface of the well.
c. In spare wells surrounding those with scaffolds, PBS was added to prevent the scaffolds from drying out, and then the scaffolds were incubated at 37 °C for 2 h to allow cells to attach to the scaffold.
3. Post-seeding culture (1–21 days)
a. Following 2 h of incubation, 500 μl complete medium was added to the individual wells to submerge the scaffold by gently pipetting the media down the side of the well. Scaffolds were then incubated at 37 °C, 5% CO2 until required for the next experimental step.
b. Where necessary, media should be replaced every 3–4 days by careful pipetting to remove old media and then replaced with fresh media.
NOTE: Since myeloma cells grow in suspension, direct adherence to the scaffold is variable and influenced by the presence of osteoblasts and the success of seeding as a single droplet. Nevertheless, droplet-based seeding was effective for achieving distribution throughout the scaffold pores.
NOTE: The total media volume added to wells could be increased to 1 ml total if needed, ensuring the media is gently and slowly added down the well wall.
Several methods can be used to process the scaffolds for analysis. Below are examples of how we processed the scaffolds; however, this is not an exhaustive list.
1. Dissociation of cells from scaffolds (e.g. for flow cytometry)
a. The scaffolds were removed from the wells and cultured in fresh 48 well plate.
b. Wash scaffolds in 400 μl PBS for 5 min at room temperature on a shaker in order to wash FBS from scaffolds and to isolate cells that are weakly attached to scaffold discs.
c. The scaffolds were moved to a new well containing 300 μl of accutase solution and incubated at room temperature on a shaker for 30 min.
d. The scaffolds were moved to a new well containing 250 μl of trypsin-EDTA and incubated at 37 °C for 10 min.
e. Quench trypsin by adding 500 μl serum containing media to wells.
f. Combine PBS, accutase, and trypsin suspensions to 1.5 ml Eppendorf tubes.
g. Scaffolds were cut using scissors or a blade into quarters and placed into an extra Eppendorf tube containing 500 μl PBS.
h. Centrifuge all tubes (from steps f and g) at 800xg for 5 minutes.
i. Remove supernatant from tubes and resuspend pellets in 100 μl PBS.
j. Live/dead staining was added to each Eppendorf tube and incubated at room temperature for 30 min.
k. Centrifuge suspension at 600xg for 5 min and resuspend cells in PBS containing 10% FBS.
l. Run samples on a flow cytometer, gating for live/viable populations, and for DiD staining profiles.
2. Scaffold fixation
a. Scaffolds were removed from the wells post-culture and placed into fresh wells of a 48 well plate.
b. Gently wash scaffolds with 500 μl PBS for 10 minutes at room temperature.
c. Remove the PBS and repeat the above step a further two times (a total of three washes should be performed).
d. Remove PBS and add 4% PFA to wells, incubate at room temperature for 15 min.
e. PFA was removed and the scaffolds were washed in 500 μl PBS for 10 min at room temperature.
f. Remove the PBS and repeat the above step a further two times (three total washes should be performed).
g. PBS was removed and 500 μl DAPI solution (DAPI:PBS ratio 1:1000) was added to the wells, followed by incubation at room temperature for 10 min.
h. The DAPI solution was removed, and the scaffolds were washed in 500 μl PBS for 10 min at room temperature.
i. Remove the PBS and repeat the above step a further two times (three total washes should be performed).
j. The scaffolds were maintained in PBS at 4 °C and wrapped in foil until sectioning was performed (up to a month).
NOTE: To maintain representative architecture of cell growth, the scaffolds were handled carefully and reagents added gently to wells to minimise the disruption of cells on the scaffolds.
3. Scaffold frozen sectioning
a. A cryostat chamber should be set to −20 °C.
b. Prepare a cryo-mold by placing enough OCT to fill the cryo-mold, ensuring that no bubbles are present in the OCT layer, as this may cause cracking of the OCT and scaffold in later steps.
c. Previously fixed scaffolds (Protocol 5, step 2) were removed from the wells containing PBS, and excess liquid was removed by dabbing the edge of the scaffold onto a paper towel.
OPTIONAL: Scaffold can be cut in half to create 2 equally sized semi-circles or left as a whole disc.
d. The scaffold was then dipped in OCT and placed in the prepared mold. If the scaffold was cut, each half was placed such that the straight edge was facing down. If the scaffold was not cut, it was placed flat on OCT.
NOTE: Each method produces scaffolds that are embedded in a different plane. This can also be achieved by sectioning the OCT block at different angles (see below). We suggest the scaffold should be kept whole as it allows better control of the positioning of the scaffold in the mould. If time to section a scaffold is the limiting factor, it is possible to place the semi-circles flat with one laid on top of the other, but once again this gives the user less control over the scaffolds.
e. The OCT mold was immersed in an ethanol/dry ice bath or liquid nitrogen for 10–15 seconds, and then quickly transferred to the cryostat chamber.
f. The solidified OCT block was removed from the mold and attached to a steel chuck in the required orientation by placing a drop of OCT onto a cooled steel chuck and then placing the OCT block onto it.
g. The OCT block was allowed to stick to the chuck by placing the chuck in the cryostat chamber for 5 min or until the freshly laid OCT solidified.
h. Cut into the block until the scaffold is located.
i. Take sections of the scaffolds at 10 μm to 20 μm thickness.
j. The section was flattened out using a brush and collected onto a glass slide by placing the slide over the cut section.
k. Allow sections to air dry on scaffolds for 3 hours prior to imaging.
NOTE: Marking the top of the OCT mould with a pencil once it has been solidified helps the user to orientate the block.
NOTE: Using coloured OCT may help with the identification of the scaffold rather than white OCT since the scaffolds are also white.
NOTE: Aqueous mounting media can be used to fix a coverslip to the slide to preserve the sample longer.
1. Uneven cell distribution or poor seeding efficiency
Issue: Cells failed to adhere evenly or settle onto the plastic well plate instead of the scaffold.
Steps to troubleshoot:
• Ensure that the scaffolds are dry on the surface before seeding to help the droplets stay in place. This can be achieved by placing scaffolds at 37 °C if previously performed only at room temperature. Alternatively, the drying time could be increased.
• Ensure that cells were seeded in a 10–20 μl droplet directly onto the centre of the scaffold to maximise coverage.
• When incubating cells on the scaffolds initially, a pre-warmed incubator was used to avoid disturbing the plates during the initial 2-hour attachment period.
• Scaffolds were moved to new wells to avoid the attraction of cells to those adhered to the well plate plastic rather than the scaffold.
2. Scaffolds floating on top of media layer in well plates
Issue: Scaffolds float because of disruption during media addition or incubation, resulting in poor cell contact or movement in the well.
Steps to troubleshoot:
• Add medium slowly down the side of the well after the initial 2-hour seeding phase.
• Avoid agitation during media changes and move plates gently during transportation.
• If scaffolds float after the addition of media, gently press the scaffold to the bottom of the well using the pointed end of a 200 μl sterile plastic pipette tip.
3. Loss of non-adherent cells (e.g. myeloma)
Issue: Suspension cells are lost or do not adequately attach to the scaffold.
Steps to troubleshoot:
• Cells were allowed to settle by initially seeding in a smaller volume (10 μl) before adding larger volumes of media.
• Handle plates gently to avoid agitation of the scaffold.
• The frequency of media changes was reduced to minimise the disruption of myeloma cells on scaffolds by increasing the total well volume to 1 ml.
• Monitor suspension cells in supernatant as part of overall viability readouts.
4. Low cell recovery from scaffolds
Issue: Insufficient cell yield from digested scaffolds for flow cytometric or molecular analyses.
Steps to troubleshoot:
• Optimise enzymatic digestion protocols (e.g., use of collagenase/accutase or increased incubation length). This will be cell dependent, but the use of accutase at 30 min, followed by trypsin for 5 min, has shown no effect on cells in our hands.
• Use agitation (e.g. plate shaker) and centrifuge scaffolds to improve recovery.
• Pool multiple scaffolds for endpoint analyses that require higher cell input (e.g., RNA extraction).
5. Imaging difficulties due to scaffold autofluorescence
Issue: Fluorescent (confocal) images are unclear due to scaffold properties.
Steps to troubleshoot:
• Include scaffold-only controls to assess background autofluorescence.
• Avoid using fluorophores with emission in the blue range (e.g., DAPI, Alexa Fluor 405), as the scaffold autofluorescence is highest at shorter wavelengths. Therefore, prioritise using fluorophores with longer emission wavelengths (e.g., Alexa Fluor 647) to improve the signal-to-noise ratio in confocal imaging.
• Optimise laser intensity to reduce background noise.
Confocal imaging of the scaffold cross-sections confirmed that the scaffolds successfully supported the co-culture and spatial organisation of myeloma and osteoblast-lineage cells. After 21 days of co-culture of 5TGM1 and MC3T3 cells on the scaffolds, both cell types were able to establish networks throughout the scaffold structure. Myeloma cells grew in direct contact with osteoblasts (Fig. 3A), adjacent to osteoblast clusters (Fig. 3B), or near osteoblast populations without direct contact (Fig. 3C). These observations indicate that the scaffolds permit both direct contact-dependent and indirect paracrine interactions, resembling the cell-to-cell interactions observed within the endosteal niche in vivo.6,23
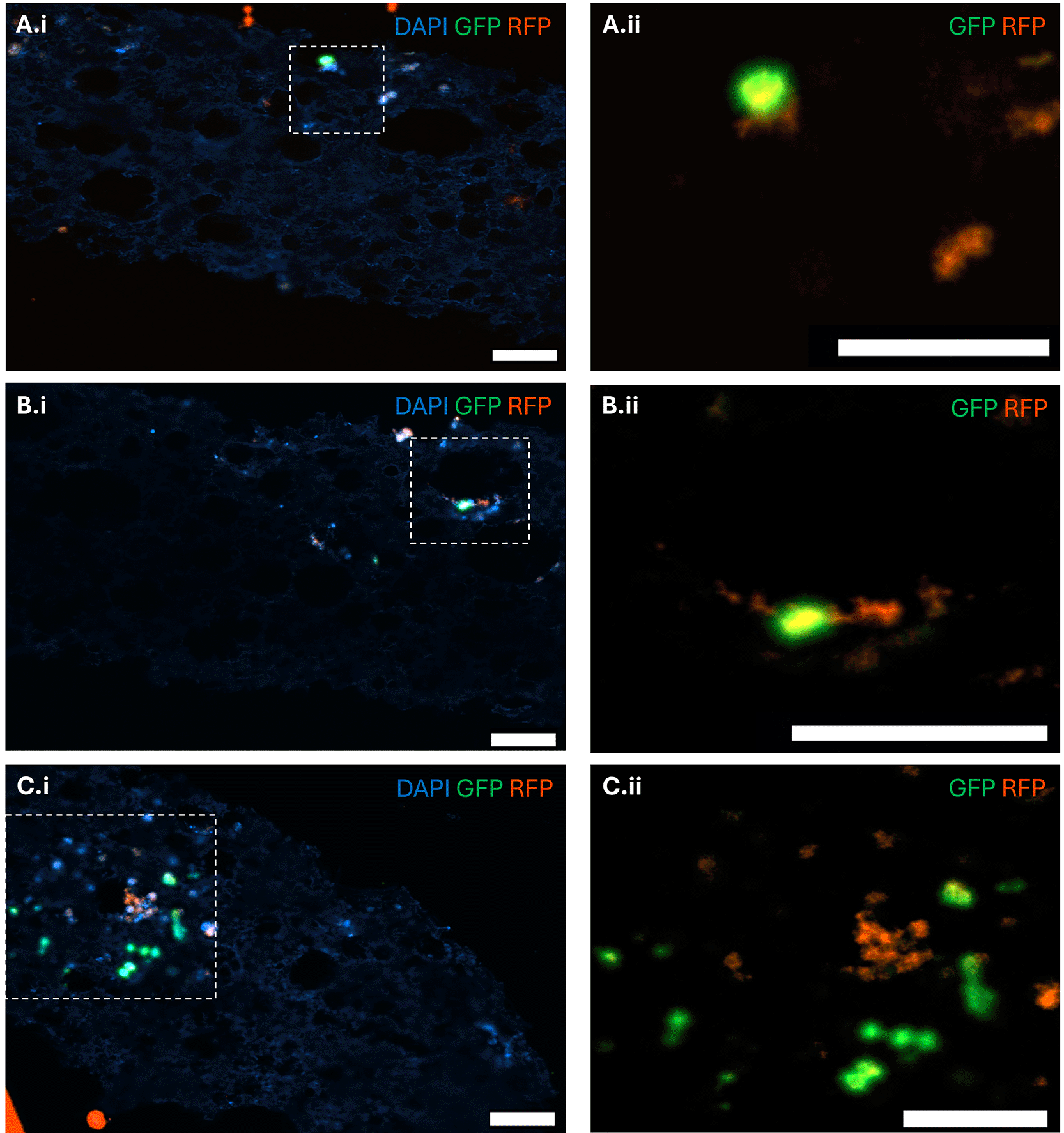
Fluorescent images of scaffold cross sections in which scaffolds were pre-seeded with RFP-tagged MC3T3 osteoblastic cells, then co-cultured with GFP-tagged 5TGM1 myeloma cells for 21 days. Scaffold structure was observed with DAPI counter stain. (A) Example of a single myeloma cell in direct contact with an osteoblast. (B) Example of a single myeloma cell neighbouring an osteoblast. (C) Example of clusters of myeloma cells and osteoblasts growing separately. Scale bar = 50 μm.
Having confirmed the co-culture architecture within our scaffolds, we next assessed drug responses on the scaffolds and compared this with standard 2D in vitro cultures. JJN-3 myeloma cells were co-cultured with either hFOB 1.19 or MG-63 osteoblastic cells and treated with 2D IC25 concentrations of melphalan, bortezomib, or lenalidomide: three standard-of-care myeloma therapies with known mechanisms of action. Whole scaffold populations were assessed using alamarBlue® assays (Fig. 4A) and showed distinct responses between the osteoblast types and culture methods (Fig. 4B). In hFOB co-cultures, cells were significantly more sensitive to melphalan on our scaffolds (3D) compared to 2D, whereas MG-63 co-cultures displayed a similar but non-significant trend. Bortezomib treatment had greater cytotoxicity in 3D MG-63 co-cultures, while hFOB co-cultures showed no difference. In contrast, lenalidomide sensitivity was reduced in 3D cultures for both osteoblast co-cultures. Despite this, for any given drug and culture format (e.g., melphalan treatment in 2D), there were no significant differences in cytotoxicity between MG-63 and hFOB co-cultures. This suggests that the sensitivity of each drug was altered by microenvironmental cues, particularly 2D vs. 3D environments, whereas the osteoblast type contributes more subtle effects that become apparent only when comparing across culture formats. Notably, alamarBlue® measures the conversion of resazurin to resorufin in all viable cells (myeloma and osteoblasts); therefore, these experiments do not allow definitive conclusions to be drawn for a specific cell type.
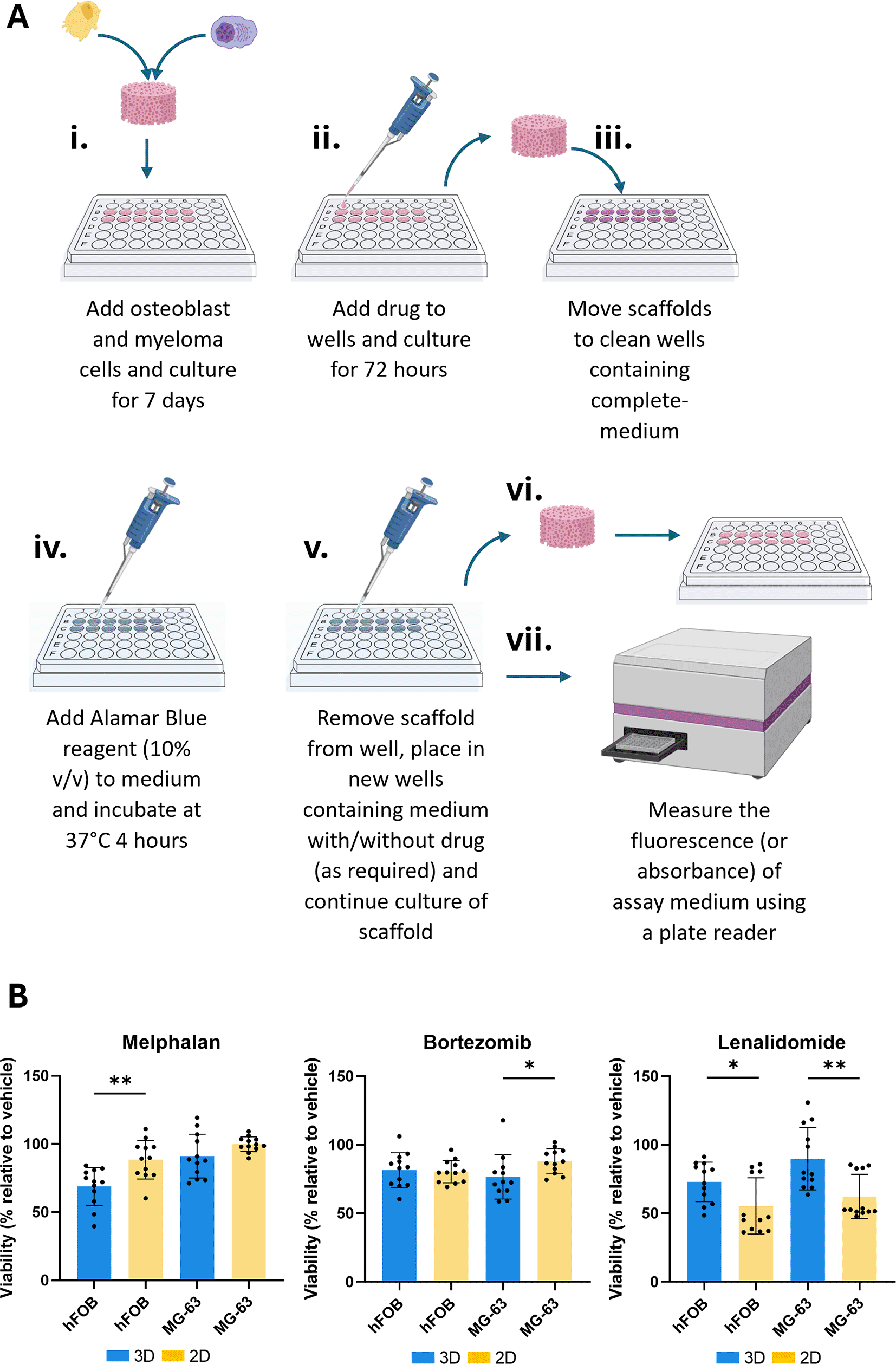
(A) Schematic of scaffold-based drug treatment workflow. Created using Biorender.com. Osteoblasts are seeded onto scaffolds, followed by myeloma cells and cultured for 7 days. Drug is then added for 72 hours. Scaffolds are subsequently transferred to fresh wells, alamarBlue® reagent is added and scaffolds are incubated for 4 hours, after which scaffolds are moved to new wells for continued culture. Fluorescence/absorbance of alamarBlue®-containing media is measured to assess cell viability. (B) Relative cell viability assessment of JJN3 cells co-cultured with hFOB 1.19 or MG-63 cells following treatment with melphalan (5.8 μM), bortezomib (1.5 nM), or lenalidomide (5 μM) in 3D scaffolds or 2D controls. Each scaffold (3D) or well (2D) was treated as a single experimental unit for statistical analysis; n = 12 experimental units per condition, comprising 3 independent runs with 4 technical replicates each. *P < 0.05, **P < 0.01.
To investigate the effects of the drugs on distinct myeloma populations, U266 myeloma cells were treated in 2D and 3D co-cultures (with hFOB or MG-63 cells) with bortezomib, lenalidomide, or a combination of both (Fig. 5). Flow cytometric analysis again showed that in 3D cultures, viability was significantly reduced following bortezomib or combination treatment compared with vehicle controls, whereas no significant differences were observed in 2D cultures (Fig. 5A-D).
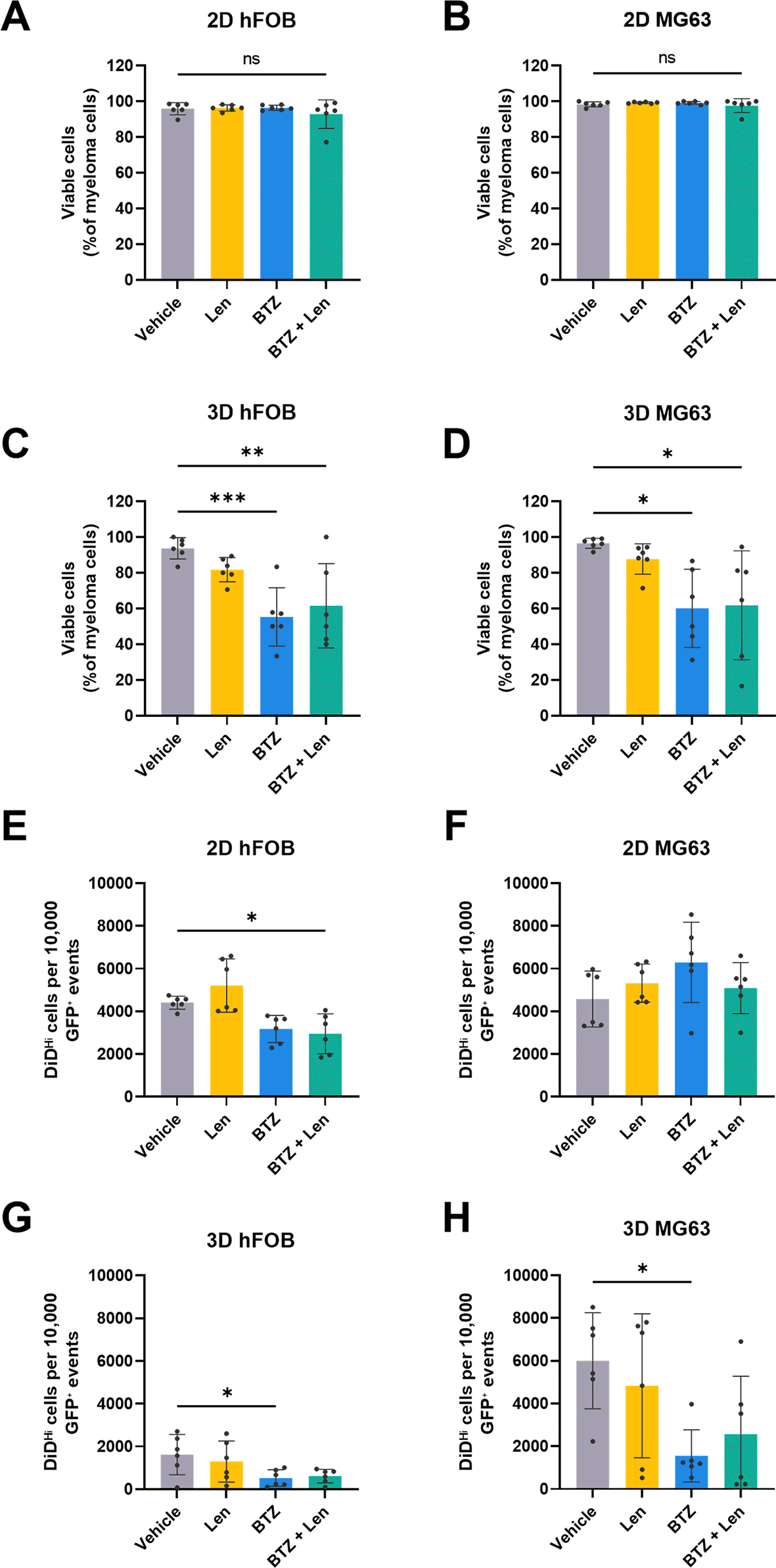
U266 cells were co-cultured with hFOB or MG-63 cells and treated with bortezomib (3 nM), lenalidomide (50 μM), or a combination of both for 72 hours. (A-D ) Cells were then retrieved from scaffolds, and the cell viability of whole U266 myeloma cell populations was assessed by live dead staining (a 2D comparison was also run under the same conditions). (E-H ) Dormant (DiDHi) U266 cell populations were also assessed following treatment. The number of DiDHi cells is presented normalised to 10,000 GFP+ cells to facilitate comparison between 2D and 3D. Each scaffold (3D) or well (2D) was treated as a single experimental unit for statistical analysis; n = 6 experimental units per condition, comprising 2 independent runs with 3 technical replicates each. * P < 0.05, ** P < 0.01, *** P < 0.01, ns P > 0.05.
When DMCs (DiDHi) were analysed separately in 2D and 3D cultures, some differences were observed. In 2D hFOB co-cultures, combination treatment led to significantly fewer DiDHi cells compared to the vehicle (Fig. 5E), while no significant differences were observed in 2D MG-63 co-cultures (Fig. 5F). However, in 3D cultures, both hFOB and MG-63 co-cultures had significantly fewer DiDHi cells in bortezomib-treated samples compared with vehicle (Fig. 5G-H), demonstrating reproducibility on our scaffolds with different osteoblastic cells, although it should be noted that fewer cells were retrieved from hFOB co-cultures on scaffolds than MG-63 co-cultures.
The polyHIPE scaffold provided a 3D, highly porous environment suitable for modelling interactions between osteoblasts and myeloma cells under physiologically relevant conditions. Its architecture supports high-resolution imaging and fluorescent tracking of proliferative and DMCs, visualisation of osteoblast networks, and co-localisation studies using confocal microscopy. Sequential seeding allows investigation of dormancy induction and osteoblast-myeloma cell interactions within a bone-lining niche-like context. The scaffold also serves as a platform for functional assays, including pharmacological testing, thereby supporting early-stage drug evaluation. Its modular design allows adaptation to different cell types, co-culture combinations, and assay formats, bridging the gap between conventional 2D culture and in vivo models, while supporting early-stage therapeutic evaluation. Below are examples of how we used some of these methods in our study.
This protocol describes the step-by-step use of a novel 3D scaffold model that replicates the key features of the BM microenvironment to study myeloma cell dormancy and drug responses. It offers a scalable, reproducible, and adaptable platform that minimises reliance on animal models in line with 3Rs principles. The validation of this model against existing in vivo data is ongoing. However, we demonstrated the compatibility of our model with human and mouse osteoblastic cells as well as human and mouse myeloma cells. We also demonstrate a range of downstream analysis methods using our model, which widens the potential applications of this model. For instance, confocal imaging allowed us to validate that the polyHIPE scaffolds were capable of recreating the spatial organisation, facilitating direct and indirect contact between osteoblasts and myeloma cells, which is characteristic of the BM niche. These interactions are important and difficult to model simultaneously in 2D in vitro systems. Thus, the ability to visualise these interactions on our scaffolds underscores their suitability for studying cell-to-cell interactions and relationships.
Through drug testing on our model compared to 2D in vitro cultures, we were able to confirm the influence of the 3D structure, which largely increased drug sensitivity. Importantly, the use of a range of cell lines in our model reduces the need for animal-derived materials and increases the adaptability of our model. Using a range of osteoblast cell lines for co-cultures, we were able to confirm the ability to derive similar conclusions across several osteoblast phenotypes, which are important to consider as they may influence therapeutic responses. The use of hFOB and MG-63 cells, which are two commonly used osteoblastic cell lines in research laboratories, was interchangeable, even with different myeloma cell lines. Importantly, the similarities observed on our scaffold when using different osteoblast types were not consistently replicated in 2D culture, highlighting the detrimental effect of 2D culture compared with 3D culture. At the cellular level, flow cytometry confirmed that DMCs could be targeted on our scaffolds, mirroring observations made previously using in vivo models 6 and emphasises our model’s capacity to study dormancy-associated resistance mechanisms. Ongoing work aims to further validate our 3D model using in vivo data. We hope to further explore the integration of patient-derived samples to enhance the clinical relevance of the model.
Compared with other 3D models, our polyHIPE scaffold offers several practical and biological advantages. Hydrogel-based or collagen scaffolds often lack the structural rigidity and pore interconnectivity needed for long-term co-culture or imaging, while microfluidic “bone marrow-on-a-chip” systems are technically complex and difficult to scale for parallel experiments. PolyHIPE scaffolds bridge this gap, offering a comparatively higher-throughput system that combines stability, tunability, and optical accessibility with compatibility for imaging, biochemical, and flow cytometric analyses. Thus, it is a suitable model for preclinical application. Importantly, the model aligns with NC3Rs 3Rs principles. By reproducing key aspects of the BM microenvironment in vitro, the scaffold enabled the evaluation of cellular dormancy and drug responses without relying on murine models.
Based on the work presented here, we suggest that scaffold-based assays could replace small pilot in vivo studies, typically with ~10–20 animals per study. Widespread adoption of this model has the potential to reduce the use of hundreds of animals annually across the field, minimising the experimental burden of research while maintaining translational relevance.
Finally, the adaptability of the model extends its potential beyond myeloma. By varying cell types, ECM coatings, or scaffold composition, it can easily be adapted for studying broader aspects of myeloma (aside from dormancy) or for studying other cancers such as breast or prostate dormancy. Adoption by other researchers can be aided by sourcing pre-made scaffolds from our in-house facility; however, the potential to manufacture the scaffolds within separate laboratories requires specialised equipment (e.g., vibratome), which could limit the uptake of the model.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)