Introduction
The epigenetic regulation of gene expression1 has recently attracted the interests of many researchers. Epigenetic modifications regulate gene expression without modifying DNA sequences. Examples include promoter methylation2, histone modification3, the binding of transcription factors to gene promoter regions4, and miRNA regulation of target genes5.
Promoter methylation and miRNA regulation of target genes are particularly important in the epigenetic regulation of gene expression. Promoter methylation is relatively stable, long term, and in some cases, heritable. It is generally believed that genes with hypermethylated promoters are repressed. In addition, there is mounting evidence that DNA methylation is involved in the development and progression of certain disease states. For example, aberrant methylation in cancer is frequently observed6 and the distinct patterns of promoter methylation between monozygotic (MZ) twin pairs have also been found to result in different health conditions7. In contrast to DNA methylation, miRNA regulation of target genes is more flexible and can change even during cellular differentiation8. miRNA expression is often tissue-specific, and similar to DNA methylation, miRNA expression has been linked to human disease9. Thus, although miRNA-directed gene regulation is thought to result in subtle changes, it is generally believed that miRNAs are involved in many important biological processes ranging from cell division to aging.
Although DNA methylation of miRNA promoters has been studied extensively (e.g., with respect to tumor formation10), the relationship between promoter methylation and miRNA regulation of target genes has not been thoroughly investigated. One likely reason for this is that the regulation of gene expression by promoter methylation is a form of pre-transcriptional control, whereas miRNA regulation of target genes is a form of post-transcriptional control, with the former taking place inside the nucleus and the latter outside the nucleus (cytoplasm). Thus, these two mechanisms are separated by both time and space, and as a result, there have not been plausible biological reasons to suspect that promoter methylation and miRNA-mediated gene regulation operate in concert.
However, Su et al.11 recently found that miRNAs have a tendency to target genes with hypomethylated promoters. To my knowledge, their study was the first report suggesting coregulation of gene expression by promoter methylation and miRNAs. In addition, Sinha et al. reported that genes promoters with high CpG content were more often targeted by miRNAs12. Saito and Sætrom also discussed the relationships between miRNA-mediated gene regulation and various features of target genes, but they did not consider the methylation status of target gene promoters13. Although the study of Su et al represents the first evidence of a direct link between promoter methylation and miRNA regulation, the biological significance of their findings is not clear. In this study, I report that promoter methylation is associated with miRNA-targeting; that is, the amount of methylation observed at a given gene promoter is dependent on whether that gene is also a target of miRNA regulation. Furthermore, miRNA-targeted-gene promoter methylation is also related to how miRNAs regulate target gene expression. In particular, I reveal that miR-548 miRNAs target genes are associated with highly hypomethylated promoters.
Methods
Promoter methylation profiles
In this study, I used publically available promoter methylation profiles from various resources obtained from GEO ID: GSE3065314. This included 283 human promoter methylation profiles for distinct cell lines, ranging from hESC to various somatic samples, measured using the HumanMethylation27 BeadChip (Illumina), which provides an efficient approach for surveying genome-wide DNA methylation profiles. The HumanMethylation27 panel targets CpG sites located within proximal promoter regions of transcription start sites (TSS). Thus, it was suitable for the purpose of this study. Promoter methylation profiles (GEO ID: GSE30653) also included data from both IMR90 and MRC5 cell lines, which were used to investigate relationships between promoter methylation and previously reported miRNA regulation and miRNA expression profile data15,16. Promoter methylation profiles in both BG02 and BG03 were also included in this study, and were compared to miRNA regulation and miRNA expression profile data (see below).
Additional promoter methylation profiles in IMR90 cell lines were obtained from GEO IDs, GSM86800814, GSM73994017, and GSM37544218. They were compared to IMR90 promoter methylation profiles (GEO ID: GSM760387 within GSE30653). GSM868008 was included in the GEO ID, GSE31848, which were generated using the Illumina HumanMethylation450 BeadChip. This BeadChip allowed us to interrogate > 485000 methylation sites per sample at single-nucleotide resolution. In addition, because this array also includes CpG sites outside of promoter regions, I restricted probes to a subset labeled as either TSS200 or TSS1500. Data from GSM739940 includes IMR90 promoter methylation profiles measured by the Illumina HumanMethylation27 BeadChip. However, because these data were generated by a different research group than that of GSE30653, I tested profiles from this dataset to confirm that obtained results were not research group dependent. Finally, I also used methylation profile data from GSM375442, which were generated using next generation sequencing (NGS). CpG methylation profiles from promoter regions were extracted using Bismark Software (Ver. 0.7.4)19 (see below); promoter regions were defined as nucleotide positions between -200 and +1200 basepairs (bp) from transcription start sites (TSSs).
mRNA and miRNA expression profiles
In order to compare miRNA-targeted-gene promoter methylation with target gene miRNA regulation and miRNA expression profile data from BG02 and BG03 cell lines, both miRNA and mRNA profiles were obtained from GEO ID: GSE1447320. Gene (mRNA) expression profiles of undifferentiated and differentiated BG02 and BG03 cell lines were obtained from the GEO IDs, GSM551204 and GSM551206, and GSM551216 and GSM551218, respectively. Corresponding miRNA expression profiles of these two cell lines were obtained from the GEO IDs, GSM361147 and GSM361271 (BG02) and GSM361288 and GSM361289 (BG03). Raw data files were downloaded for further analysis and were normalized so as to have a mean of 0 and a variance of 1.
Investigation of miRNA-targeted-gene promoter methylation
In order to infer miRNA-targeted-gene promoter methylation, I employed the MiRaGE method21 (see below). The MiRaGE method, which was implemented on a public domain MiRaGE server and Bioconductor MiRaGE package, was first used to infer the contribution of miRNA to the measured expression levels. This software was designed to accept expression profiles of the target genes in question; however, in this study, I used this method to infer miRNA-targeted-gene promoter methylation by substituting gene expression profiles with the promoter methylation profiles of each gene.
I first prepared a control dataset (pseudo) in which the expression level of all genes assigned a value of 1. Then, the amount of methylation at each gene was used as values of treatment data set. Although, the ratio of the number of methylated sites to the total number of methylated and non-methylated sites is typically used to describe promoter methylation levels, I employed a method in which total methylation values were used. I used this approach because I found that P-values computed when using methylation data were more strongly correlated to the P-values calculated from target gene miRNA regulation data (see below), which is likely due to the fact that the frequency of CpGs is also related to miRNA targeting12; i.e., genes with promoters that contain more CpGs were more often targeted by miRNAs as mentioned above. Using this procedure, I attributed two P-values to each miRNA, one expressing the degree of promoter hypermethylation, and the other expressing the degree of promoter hypomethylation. The approach used to compute P-values representing promoter methylation are described below for each of the different methodologies and/or datasets used.
GSM868008
Promoter methylation profiles used to replace “gene expression” values within the MiRaGE method were
where M0i represented the scaled values of signal_B (intensity estimated of methylated DNA), which were expressed as the amount of promoter methylation of ith gene,
where N was the total number of genes considered and Mi was the raw value of signal_B. This signified that the amount of promoter methylation was scaled so as to have a mean 〈M0i〉 of 0 and a standard deviation σM0i of 1. exp was applied in this instance because I wanted to consider the amount of methylation rather than the ratio of methylation. Because P-values where computed after the pair of input values where transformed to a logarithmic ratio, substituting 1 in the control dataset and an exponential value in the treatment dataset based on raw values resulted in the usage of raw values of differential expression/promoter methylation (see below).
GSE30653
Promoter methylation profiles used to replace “gene expression” values within the MiRaGE method were
where Ci took on the value of 1 only when Mi = 0; otherwise, it took on the value 0, so as to avoid infinite values after transformation to the logarithmic ratio.
GSM739940
Promoter methylation profiles used to replace “gene expression” values within the MiRaGE method were
where βi was the ratio of methylated sites to unmethylated sites,
where Ui was the signal from unmethylated sites (signal_A) and C was the regulation constant, which typically took the value of 100. Since only β values were deposited in the public datasets used, their use could not be avoided; however, as a result, the correlation with target gene miRNA regulation was substantially decreased. An explanation for this is noted above.
GSM375442
Promoter methylation profiles used to replace “gene expression” values within the MiRaGE method were
where max(Mi) was the maximum value of Mi and Mi was computed in this case as follows:
where yj, 0 ≤ yj ≤ 100 was the percentage of methylation at site j, which was computed using the Bismark Software19 (see below). The summation was taken over the length of the promoter region as defined above (i.e., between -200 bp and +1200 bp from the TSS).
Methylation computation by Bismark Software
The following command line inputs were used to generate methylation values of CpG sites within the Bismark Software package19.
% bismark_genome_preparation \
–path_to_bowtie bowtie_dir \
–verbose./hg19/ &
% R
>x <- scan("GSM375442_CpgMIP-IMR90.seq.txt",
sep="\n",what=character(0))
>write.table(file="sequence.fa",
paste(paste(">p",1:length(x),sep=""),
x,sep="\n"),sep="\n",row.names=F,
quote=F,col.names=F)
>q()
% bismark,/hg19/ \
–path_to_bowtie bowtie_dir \
–bowtie2 -f sequence.fa
% methylation_extractor -s –comprehensive \
sequence.fa_bt2_bismark.sam
% genome_methylation_bismark2bedGraph_v3.pl \
CpG_content_sequence.fa_bt2_bismark.sam.txt \
> sequence.fa_bt2_bismark.sam.bed
where bowtie_dir is the directory where bowtie2 is installed. In this study R22 was used, but scripts executed by R can also be perfomed by any other alternative languages. GSM375442_CpgMIP-IMR90.seq.txt is a file downloaded from GEO and sequence.fa_bt2_bismark.sam.bed includes methylation percentages of each CpG site, yj, which was explained above.
Inference of miRNA-targeted-gene promoter methylation/miRNA regulation of target genes using the MiRaGE method
The inference of miRNA-targeted-gene promoter methylation/miRNA regulation of target genes was carried out using the MiRaGE method, which has been described previously21.
Although the MiRaGE method is typically used for datasets with two experimental conditions, each of which contains more than one replicate, I used this method for instances in which each condition consisted of only a single replicate. In each case, I based the analysis on the premise that for a given gene i, there were a pair of gene expression datasets or promoter methylation profile datasets, which were measured under a control condition xcontrol,i and a treatment condition xtreat,i. From this, I computed the logarithmic ratio
In such an example, when a difference between raw values is favorable, exponential values exp(xi) can be used instead of xi, in which case, I got
When computing P-values that rejected the null hypothesis, by using the alternative hypothesis that ∆xis of the target genes of the miRNA m are less (greater) than those that are off-target but form a target of other miRNAs, I computed
where P[A<(>)B] represented P-values computed by statistical tests when sets A and B are compared. The tests implemented within the MiRaGE Server/package are the t-test, Wilcoxon rank sum test, and Kolmogorov-Smironov test.
Thus, determining whether A < (>)B depends on the selected statistical test used. I used Gm to represent the set of genes targeted by miRNA m and G’m as the intersection of the set of off-target genes of miRNA m and the set of target genes of all other miRNAs. It should be noted that genes that were not the targets of any miRNAs were totally excluded from the analysis; however, all miRNAs were considered and no exclusions based on conservation were applied. When inferring promoter methylation, xcontrol,i = 1 and xtreat,i were used to represent the amount of promoter methylation. When inferring miRNA regulation of target genes during cell senescence in IMR90 and MRC5 cell lines, xcontrol,i was used to represent gene expression of young cell lines and xtreat,i was used to represent gene expression in senescent cell lines. When inferring regulation of target genes during differentiation in BG02 and BG03 cell lines, xcontrol,i was used to represent gene expression in undifferentiated cell lines and xtreat,i for gene expression in differentiated cell lines.
Correlation between miRNA-targeted-gene promoter methylation and miRNA regulation of target genes
I used two types of P-values, P<(>)m:methy, which corresponded to the miRNA-targeted-gene promoter methylation, and P<(>)m:regul, which corresponded to the miRNA regulation of target genes of miRNA m. When P<(>)m:methy[regul] was small enough, the target genes of miRNA m are significantly hypermethylated or hypomethylated and thus downregulated or upregulated. In order to see if these two types of P-values were correlated, I computed various correlation coefficients:
and accompanied P-values to reject null hypothesis that ρ = 0 by using the alternative hypothesis that ρ ≠ 0. I used ρ[a, b] as the Pearson’s correlation coefficients between a and b and rank(xm) as the rank order of xm among {xm}. Pm:z where z ∈ {methyl, regul} was either P<m, P>m, 1 – P<m or 1 – P>m. Thus, there were 4 × 4 = 16 possible combinations of Pm:methy and Pm:regul. ρs of the Kolmogorov-Smirnov test and ρlogPearsons for all tests changed when P<(>) was replaced with 1 – P>(<) because P<(>) ≠ 1 – P>(<) for the Kolmogorov-Smirnov test and log(P<(>)) ≠ log(1 – P>(<)) for all tests. Thus, optimal combinations of the maximum absolute correlation coefficients were employed.
The reciprocal relationship between miRNA regulation of target genes and miRNA expression during differentiation in BG02 and BG03
In contrast to the cell senescence study15 in which miRNA expression was investigated by NGS, only microarray measurements were available for differentiation in BG02 and BG03 cell lines. Due to issues related to the accuracy and quality of microarray data and the relatively small amount of miRNA expression, very few miRNAs were found to be differentially expressed between undifferentiated and differentiated cell lines. Thus, miRNAs that were differentially expressed between undifferentiated and differentiated cell lines were selected based on two criteria before being subjected to further analyses:
Absolute differential expression | xdiff,i – xundiff,i |> ∆xc, where xdiff,i and xundiff,i were the normalized expression of gene i of differentiated and undifferentiated cell lines, respectively. ∆xc was set as the threshold value that could be used to select genes associated with significant differential expression during differentiation.
For this method, it was required that adjusted P-values based on the BH criterion23 were less than 0.05 for significant upregulation or downregulation. Here, P-values were computed using a t test between two sets of probe values attributed to gene i.
After ∆xc was suitably selected, the correlation coefficient between differential expression xdiff,i – xundiff,i and log (P<m:regul) were computed. Positive values were taken to indicate a reciprocal relationship between miRNA expression and miRNA regulation of target genes; smaller P<m:regul were assumed to signify that target genes were upregulated (and vice versa). Thus, a reciprocal relationship required that miRNA were downregulated, i.e., xdiff,i – xundiff,i <(>)0, which should result in a positive correlation between xdiff,i – xundiff,i and log (P<m:regul). Since miRNA names in GSE14473 were old (miRBase, release 9.1), they were converted to miRNA names used in the present version of the MiRaGE software package (miRBase release 18) and by the miRConverter implemented in miRSystem24.
Ranking of miRNAs having target genes with miRNA-targeted-hypomethylated promoters
miRNAs were ranked based on P>m in each of the 283 samples in GSE30653, after excluding 12 control samples. Each miRNA was ranked within each sample, and then, the cumulative rank was determined for all 270 samples.
Ranking of miRNA regulation of target genes
miRNAs were ranked based on P-values for either downregulation during cell senescence (IMR90 and MRC5) or upregulation during differentiation (BG02 and BG03) for each statistical test. Then, each miRNA was ranked based on the order summed up over three statistical tests.
Comparison of promoter methylation between miRNA-target genes and miRNA-non-target genes
I compared promoter methylation between miRNA-target genes and miRNA-non-target genes for each of the 270 samples in GSE30653, excluding 12 control samples. Statistical tests used were the two-sided t test, Wilcoxon rank sum test, and Kolmogorov-Smirnov test, resulting in 270 P-values for each test.
P-value computation for miRNA-targeted-gene promoter methylation after random permutation
In order to see if P-values for miRNA-targeted-gene promoter methylation, P>m, changed after the random gene permutation, gene IDs were randomly permuted for each of the 270 samples. Then, the number of significant miRNAs whose target gene promoter was significantly hypomethylated (P-values computed with t test and corrected using the BH criterion23 were less than 0.05) was counted. The numbers were averaged over one hundred independent random permutations.
Significance of correlation between miRNA-targeted-gene promoter methylation and miRNA-mediated regulation of target genes after random gene permutation
In order to see if the correlation between miRNA-targeted-gene promoter methylation and miRNA-mediated regulation of target genes changed after random gene permutation while conserving the correlation between promoter methylation and gene expression, promoter methylation and gene expression profiles needed to be permuted simultaneously. However, because gene expression and promoter methylation were measured by distinct microarrays, the number of probes attributed to each gene in some cases differed between the microarray used for gene expression measurement and promoter methylation. Also, multiple CpG sites may be attributed to each gene, while gene expression is unique to each gene. In order to overcome this difficulty, multiple probes or CpG sites were first grouped based on overlapping RefSeq mRNA IDs. RefSeq gene IDs were then shuffled and P-values were computed using MiRaGE, which employs a t test for both miRNA-targeted-gene promoter methylation and miRNA-mediated regulation of target genes, before and after random permutation. The methylation data sets used were GSM868008 (for IMR90 cell line) and GSE30653 (for MRC5, BG02 and BG03). ρPearson were computed with Pm:methy = P<m and Pm:regul = P<m. Gene expression profiles reported previously for both IMR90 cell lines and MRC5 cell lines were used15. The maximum and minimum values of ρPearsons among 100 independent random permutations are presented. The geometric average for over 100 random permutation of P-values attributed to ρPearson was also computed. In addition, P-values associated with ρPearson before random permutation was estimated based on 100 random permutation ensembles assuming a normal distribution. The normality of the distribution of correlation coefficients from 100 random permutations was checked using both the Shapiro-Wilk normality test and the Kolmogorov-Smirnov test (no significant differences from a normal distribution were detected).
Results and discussion
Significant promoter hypomethylation of miRNA target genes
Based on the inference of data produced using the MiRaGE method21 (see Methods), the promoters of genes that are targets of 70–90% of human miRNAs were significantly hypomethylated, dependent on the statistical tests used, and the definition of promoter methylation levels: the β-value or the amount of methylation (see Table 1 and Additional files). This finding was consistent with conclusions made by Su et al.11, who stated that miRNAs had a tendency to target genes with hypomethylated promoters. Given that promoter methylation patterns do not change drastically between certain cell types, the amount of miRNA-targeted-gene promoter methylation is also highly cell-type-independent. Mean correlation coefficients of P>m ranged between 0.8 and 0.9, again depending on the statistical test used and the definition of promoter methylation levels (Table 2).
Table 1. miRNA-targeted-gene promoter hypomethylation.
|
Amount of methylation
|
|---|
|
Statistical tests
|
Number of miRNAs
| |
|---|
|
Not significant
|
Significant
|
|---|
|
t-test | 136054 | 384537 | 74% |
| Wilcoxon rank sum test | 144572 | 376019 | 72% |
| Kolmogorov-Smirnov test | 129998 | 390593 | 75% |
|
β
-values
|
|
Statistical tests
|
Number of genes
| |
|
Not significant
|
Significant
|
|
t-test | 39390 | 481201 | 92% |
| Wilcoxon rank sum test | 45188 | 475403 | 91% |
| Kolmogorov-Smirnov test | 41952 | 478639 | 92% |
Table 2. Mean correlation coefficients between P-values of miRNA-targeted-gene promoter hypomethylation of samples.
Mean Pearson’s correlation coefficients of P>m between pairs of cell lines.
|
Statistical test
|
Averaged correlation coefficients
|
|---|
|
Amount of methylation
|
|---|
|
t-test | 0.9112043 |
| Wilcoxon rank sum test | 0.8981256 |
| Kolmogorov-Smirnov test | 0.8263153 |
|
β
-values
|
|
t-test | 0.8919359 |
| Wilcoxon rank sum test | 0.8832241 |
| Kolmogorov-Smirnov test | 0.8257556 |
In order to confirm that the observed miRNA-targeted-gene promoter methylation was really caused by miRNA targeting, I compared promoter methylation of miRNA target genes with that of miRNA non-target genes. In order to check this point, I have computed P-values that compare promoter methylation between miRNA target genes and miRNA non-target genes (Table 3). I found that the difference was strongly significant. P-values were clearly small enough to be significant even if any multiple comparison corrections were taken into account. This significant difference observed here is coincident with the findings of Su et al.11 and supports the argument that the observed miRNA-targeted-gene promoter methylation was really mediated through miRNA targeting.
Table 3.
P-values that represent the difference of promoter methylation between miRNA target genes and miRNA non-target genes.
| t test | Wilcoxon rank sum test | Kormogov-Smirnov test |
|---|
| Minimum | Maximum | Minimum | Maximum | Minimum | Maximum |
|---|
| 1.38 × 10–60
| 4.66 × 10–25
| 4.26 × 10–105
| 1.56 × 10–50
| 0 | 0 |
It may seem strange that I did not investigate promoter hypermethylation and hypomethylation distinctly, because these two should have different biological mechanisms. However, there are some cases that miRNA-mediated regulation of target genes is known to function toward opposite direction when third players take place. For example, some miRNAs can remove Ago proteins that bind other miRNAs25. In such cases, target genes of these miRNAs are assumed to be upregulated, as miRNAs are generally believed to suppress the expression of their target genes. miRNA sponges can also inhibit miRNA suppression of target genes26. miRNA sponges absorb miRNAs that bind to the RNA-induced silencing complex (RISC). Because of this effect, the expression of miRNAs targets tends to increase with increasing concentrations of miRNA sponges. The biological mechanisms that mediate the interactions between miRNA and methylated promoters, and the role of third players are unknown, thus, it is not always necessary to distinguish the mechanisms underlying the hyper- and hypomethylation of miRNA target genes.
Another possible issue is that miRNA-targeted-gene promoter methylation may represent an artefact caused by the complex algorithm employed by MiRaGE. In order to deny this possibility, I computed miRNA-targeted-gene promoter methylation after random permutation (Table 4), from which I determined that there was no significant miRNA-targeted-gene promoter methylation. Thus, it is apparent that random permutation destroys miRNA-targeted-gene promoter methylation completely. This demonstrates that miRNA-targeted-gene promoter methylation is not an artifact but a real outcome.
Table 4. miRNA-targeted-gene promoter hypomethylation after random gene permutation.
| Amount of methylation |
|---|
| Statistical tests | Number of miRNAs | |
|---|
| Not significant | Significant |
|---|
| t-test | 520585 | 6 | 1.1 × 10–3 % |
| Wilcoxon rank sum test | 520588 | 3 | 5.8 × 10–4 % |
| Kolmogorov-Smirnov test | 520585 | 6 | 1.1 × 10–3 % |
Significant correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation
In order to see if miRNA-targeted-gene promoter methylation was genuinely related to the miRNA regulation of target genes, I compared P-values of miRNA-targeted-gene promoter methylation to P-values of miRNA regulation of target genes during the senescence of IMR90 and MRC5 cell lines15,16 and during the differentiation of BG02 and BG03 cell lines20. It was clear that promoter methylation and miRNA regulation of target genes were significantly correlated during both cell senescence and differentiation (Table 5), despite the fact that correlation coefficients exhibited opposite directionalities in cell senescence and differentiation. This means that genes with miRNA-targeted-gene promoter hypomethylation are downregulated during cell senescence, but upregulated during differentiation.
Table 5. Correlation coefficients between P-values of miRNA-targeted-gene promoter hypomethylation and miRNA regulation of target genes.
| Statistical test | ρPearson | ρPearsonlog | ρSpearman |
|---|
|
Cell senescence
| Pm:methy = P>m vs. Pm:regul = P>m |
| IMR90 |
|
t test | 0.33 | 0.40 | 0.41 |
|
P-values | * | * | * |
| Wilcoxon rank sum test | 0.28 | 0.35 | 0.33 |
|
P-values | * | * | * |
| Kolmogorov-Smirnov test | 0.31 | 0.72 | 0.61 |
|
P-values | * | * | * |
| MRC5 |
|
t test | 0.22 | 0.23 | 0.25 |
|
P-values | * | * | * |
| Wilcoxon rank sum test | 0.53 | 0.76 | 0.73 |
|
P-values | * | * | * |
| Kolmogorov-Smirnov test | 0.38 | 0.82 | 0.67 |
|
P-values | * | * | * |
|
Differentiation
| Pm:methy = P>m vs. Pm:regul = 1 – P<m |
| BG02 |
|
t test | -0.25 | -0.68 | -0.67 |
|
P-values | * | * | * |
| Wilcoxon rank sum test | -0.13 | -0.52 | -0.53 |
|
P-values | 2.4 × 10–9
| * | * |
| Kolmogorov-Smirnov test | -0.16 | -0.70 | -0.69 |
|
P-values | 2.8 × 10–12
| * | * |
| BG03 |
|
t test | -0.31 | -0.57 | -0.53 |
|
P-values | * | * | * |
| Wilcoxon rank sum test | -0.14 | -0.23 | -0.23 |
|
P-values | 8.9 × 10–11
| * | * |
| Kolmogorov-Smirnov test | -0.14 | -0.21 | -0.19 |
|
P-values | 1.3 × 10–10
| * | * |
To confirm that the significant correlation noted above between miRNA-targeted-gene promoter methylation and miRNA regulation of target genes was not a false-positive finding, I also tested for this association in IMR90 cell lines by using a different microarray and NGS approach, as described previously18 (Table 6). Again, I observed a significant correlation between promoter methylation status and miRNA regulation, suggesting that the relationship found between these two mechanisms was independent of experimental condition.
Table 6. Evaluation of the correlation between miRNA-targeted-gene promoter methylation and miRNA regulation of target genes.
| Statistical test | ρPearson | ρPearsonlog | ρSpearman |
|---|
| GSM868008 | Pm:methy = P>m vs. Pm:regul = P>m |
| t test | 0.28 | 0.37 | 0.37 |
| P-values | * | * | * |
| Wilcoxon rank sum test | 0.18 | 0.32 | 0.28 |
| P-values | 4.4 × 10–16 | * | * |
| Kolmogorov-Smirnov test | 0.16 | 0.70 | 0.54 |
| P-values | 1.9 × 10–12 | * | * |
| GSM739940 | Pm:methy = P>m vs. Pm:regul = P>m |
| t test | 0.11 | 0.40 | 0.34 |
| P-values | 3.0 × 10–6 | * | * |
| Wilcoxon rank sum test | 0.07 | 0.33 | 0.27 |
| P-values | 2.5 × 10–3 | * | * |
| Kolmogorov-Smirnov test | 0.07 | 0.79 | 0.51 |
| P-values | 2.7 × 10–3 | * | * |
| GSM375442 | Pm:methy = P>m vs. Pm:regul = P>m |
| t test | 0.19 | 0.17 | 0.26 |
| P-values | 3.0 × 10–6 | 1.5 × 10–14 | * |
| Wilcoxon rank sum test | 0.19 | 0.27 | 0.24 |
| P-values | 2.2 × 10–16 | * | * |
| Kolmogorov-Smirnov test | 0.28 | 0.57 | 0.50 |
| P-values | * | * | * |
However, these findings may not represent a true correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation. For example, if the observed correlation is genuine, then the correlation between gene expression and promoter methylation should differ between miRNA target genes and non-target genes. Alternatively, a correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation could simply be a bi-product of the well-documented direct correlation between gene expression and promoter methylation. These issues arise from two assumptions: (1) the correlation between gene expression and promoter methylation is strong enough to mediate a correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation; and (2) the correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation should be strongly related to, or even directly caused by, the direct correlation between gene expression and promoter methylation. However, these two assumptions do not necessarily need to be true in all cases. For example, the correlation between gene expression and promoter methylation is not always strong and the correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation could be highly significant in instances in which the relationship between gene expression and promoter methylation is weak.
In order to test the first assumption, I computed the correlation between gene expression and promoter methylation, and revealed that the correlation between gene expression and promoter methylation is much weaker than the correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation (see the column entitled “gene expression vs promoter methylation” in Table 7). This suggests that the correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation is not likely the outcome of the correlation between gene expression and promoter methylation, but possibly vice versa. Next, I considered the second assumption, which also need not always be true. From a mathematical point of view, a correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation does not have to be directly related to the correlation between gene expression and promoter methylation. The reason is as follows. It is always possible to shuffle gene order with conserving the correlation between gene expression and promoter methylation, because the correlation does not change between pre- and post-permutation if pairs of promoter methylation and gene expression can be conserved. However, permutation inevitably alters both miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation, because permutation changes the set of genes targeted by each miRNA.
Table 7. The correlation between miRNA-targeted-gene promoter methylation and miRNA regulation of target genes after random gene permutation.
| miRNA regulation vs miRNA-targeted-gene promoter methylation | Gene expression vs
promoter methylation
(P-value) |
|---|
Before random permutation
(P-value) | After random permutation |
|---|
| Minimum | Maximum | (Mean P-value) |
|---|
| IMR90 | 0.39 (2.5 × 10–13) | -0.127 | 0.133 | (0.03) | -0.016 (2.27 × 10–5) |
| MRC5 | 0.22 (1.7 × 10–7) | -0.105 | 0.128 | (0.07) | -0.003 (0.77) |
| BG02 | -0.27 (2.0 × 10–7) | -0.141 | 0.078 | (0.01) | -0.048 (9.03 × 10–8) |
| BG03 | -0.30 (2.3 × 10–14) | -0.086 | 0.148 | (0.02) | 0.021 (0.02) |
Suppose we have N genes and M miRNAs. Even if we permute gene id g in both microarrays that measure gene expression and promoter methylation simultaneously, the correlation between gene expression and promoter methylation does not change, because pairs of gene expression and promoter methylation are conserved. However, Gm and G’m are always altered, as permutation inevitably removes some genes from Gm, and genes that were not included in Gm before permutation are added to Gm after permutation. Since both miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation are computed based on the comparison between Gm and G’m, miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation must be altered by the permutation.
In spite of the above discussion, one may still argue that a correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation may not be altered or may be altered very slightly by the permutation because the modification of Gm is necessary for the computation of both miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation, even though miRNA-targeted-gene promoter methylation itself is not likely to be significant because of permutation, as demonstrated in the previous section. (i.e., see Table 4).
In order to refuse this idea, I calculated the correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation after random permutation (Table 7) and determined that random permutation drastically reduces the correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation, although it does not alter the correlation between gene expression and promoter methylation, as discussed above. This suggests that the correlation between gene expression and promoter methylation can hardly be a cause of correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation, because correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation can take either smaller or larger values while the correlation between gene expression and promoter methylation is unaltered.
All of the above findings and discussions, together with the findings and discussions in the previous section, strongly suggest that the significant correlation between miRNA-mediated regulation of target genes and miRNA-targeted-gene promoter methylation is neither an artifact generated by the complicated algorithm employed in MiRaGE, nor a bi-product of a direct correlation between gene expression and promoter methylation, but is itself a genuine biologically significant phenomenon.
Significant reciprocal relationship between miRNA expression and target gene expression
In addition to these analyses, I also confirmed a significant reciprocal relationship between miRNA expression and target gene expression. Because it is usually believed that miRNAs downregulate target gene expression, downregulated genes should be targeted by upregulated miRNAs and vice versa. In fact, the reciprocal relationship during the senescence process of IMR90 cells has been reported15,16. In this study, I observed a similar reciprocal relationship during the differentiation of BG02 and BG03 cells20 (Table 8).
Table 8. The reciprocal relationship between target gene regulation by miRNA and miRNA expression.
| Cell lines | Δxc | Correlation coefficients | P-value |
|---|
| BG02 | 0.00 | 0.18 | 1.9 × 10–4 |
| BG03 | 0.04 | 0.51 | 0.04 |
Thus, I conclude that miRNA-targeted-gene promoter methylation is not an artifact, but a genuine biological process. One may doubt this conclusion given the finding that miRNA-targeted-gene promoter methylation is cell-line-independent and miRNAs are believed to regulate target gene expression in a cell- line-dependent manner. Although this implies an apparent discrepancy, the correlation coefficients obtained, which were at most 0.7 to 0.8, indicate that miRNA-targeted-gene promoter methylation reflects, at most, 50% to 60% of miRNA regulation of target genes. Thus, the cell line specific miRNA regulation of target genes can act within the remaining 50% to 40%. In actuality, as can be seen in Table 5, the correlations between miRNA-targeted-gene promoter methylation and miRNA regulation of target genes have opposite signs dependent on the biological processes being considered. Therefore, the fact that cell-line-independent and miRNA-targeted-gene promoter methylation partially governs cell line dependent miRNA regulation of target genes is not a discrepancy. At the moment, I have no hypotheses that explain why miRNA-mediated regulation of target genes is the opposite between cell senescence and cell differentiation in spite of the fact that miRNA-targeted-gene promoters are similarly hypomethylated. However, as discussed above, the miRNA-mediated regulation of target genes is often reversed because of the existence of unobserved third players. The underlying biological processes, which are unknown at present, will be elucidated in the future.
One should understand that my objective is not to insist that miRNA up/downregulation is associated with hyper/hypomethylation of the promoter. Nor can I confirm whether miRNA mediates promoter hypermethylation or hypomethylation. To my knowledge, there are no experimental observations that illustrate how miRNA methylate target gene promoters in animal cells. However, in plants, there are some findings that mature miRNAs in nuclei can mediate DNA methylation27. There are also some reports that mature miRNAs exist in, or can be transferred to the nucleus28, though mediation of promoter methylation is not observed. Details in animal cells are not yet known. In the present study, I observed correlations between gene expression and promoter methylation from an miRNA-centric viewpoint. The biological mechanism mediating this correlation, however, is not clear. It is plausible that an unknown biological factor regulates both gene expression and promoter methylation, and that the correlation between miRNA-targeted-gene promoter methylation and miRNA regulation of target genes may simply be an outcome of indirect regulation and promoter methylation. I cannot deny this possibility, but if the correlation between miRNA-targeted-gene promoter methylation and miRNA regulation of target genes represents a real phenomena and can be found in a wide range of biological processes, there must be a biological/mechanistic underpinning of this effect.
Significant promoter hypomethylation of miR-548 miRNAs target genes
In order to better understand the biological significance of our findings regarding miRNA-targeted-gene promoter methylation, I sought to identify the specific miRNAs that target genes with hypomethylated promoters. I found that most gene promoters targeted by miR-548 miRNAs were highly hypomethylated (Table 9; here, I define miRNAs called “miR-548” as miR-548 miRNAs). At present, little is known about miR-548 miRNAs, which likely results from the fact that miR-548 miRNAs are primate-specific miRNAs (ps-miRNAs). Because of this, ps-miRNAs were not expected to play critical roles in fundamental biological processes like cell differentiation and development, and it has so far been widely assumed that such basic biological processes are conserved among mammals. This assumption has resulted in the exclusion of ps-miRNAs from consideration as candidates for basic and important biological processes. Thus, it is thought that the roles of these ps-miRNAs may be limited to primate-specific biological processes. Another reason that miR-548 miRNAs have not been extensively investigated is that they are generally expressed at low levels; miRNAs that play critical roles are thought to be highly expressed. For example, although miRBase stores the number of short reads detected by next generation sequencing (NGS) for various miRNAs, miR-548 miRNAs have at most 100 short reads; this is in comparison to more abundant miRNAs like those from the let-7 family, which are typically represented by several million reads. This makes it difficult to detect miR-548 miRNAs using microarray technology or sequencing. Actually, although there have been a few reports noting that miR-548 family members, including miR-548 miRNAs, play critical roles, these data come exclusively from quantitative PCR (qPCR) experiments29,30.
Table 9. Evaluation of miR-548 miRNAs targeting genes with miRNA-targeting-genes promoter hypomethylation.
|
Statistical Test
|
Number of miR-548 miRNAs
|
Mean rank
|
|---|
|
Within top 100 ranked
|
Total number
|
|---|
|
t test | 43 | 68 | 216.2 |
| Wilcoxon rank sum test | 43 | 68 | 214.0 |
| Kolmogorov-Smirnov test | 43 | 68 | 215.0 |
In this study, I found that promoters of genes targeted by miR-548 miRNAs were hypomethylated. Because it is generally assumed that genes with hypomethylated promoters are expressed, the fact that promoters of target genes of miR-548 miRNAs are hypomethylated may mean that, compared to genes that are targets of other miRNAs, target genes of miR-548 miRNAs are more sensitive to changes in miRNA expression. This could explain why promoters of target genes of non-expressed miR-548 mIRNAs are specifically hypomethylated.
miR-548 miRNAs represent a large set of miRNAs, with 68 family members; thus, although these miRNAs exhibit low levels of expression, the fact that there are many family members may imply that they have important biological roles. Indeed, it has been reported that potential target genes of miR-548 miRNAs play critical roles in various biological processes31,32. Since it is also suggested that they originate from transposable elements (TEs), miR-548 miRNAs exhibit high sequence similarities while maintaining significant sequence diversity31.
During senescence processes of IMR90 and MRC5 cell lines, the target genes of miR-548 miRNAs were significantly downregulated (Table 10), whereas during differentiation of the BG02 cell line, the target genes of miR-548 miRNAs were significantly upregulated (Table 10). The fact that I observed significant changes in expression of target genes without significant changes in expression of miRNAs that target these genes implicates a role of miRNA-targeted-gene promoter hypomethylation.
Table 10. Ranking of miRNA regulation of target genes.
| Cell lines | Target genes | Number of miR-548 miRNAs | Mean rank | P-values |
|---|
| Within top 100 ranked | Total number |
|---|
| IMR90 | Downregulated | 24 | 68 | 438.2 | 9 × 10–14 |
| MRC5 | Downregulated | 16 | 68 | 382.6 | 5 × 10–7 |
| BG02 | Upregulated | 24 | 68 | 427.8 | 9 × 10–14 |
| BG03 | Upregulated | 3 | 68 | 762.4 | 7 × 10–1 |
Of course, the possibility remains that there are unknown biological factors that both mediate promoter methylation of miR-548 target genes and regulate gene expression targeted by miR-548 miRNAs, and all findings here are just outcomes of this hidden biological processes. Although I cannot deny this possibility, even if there are some hidden biological processes, the fact is that miRNA-548 target genes are both methylated in promoters and their regulation is robust. Future work investigating the potential underlying mechanisms are warranted.
Conclusions
Figure 1 schematically summarises the principal findings of this study. To the best of my knowledge, this is the first time that miRNA-targeted-gene promoter methylation in many types of cell lines have been observed (Table 1 and Table 2). miRNA-targeted-gene hypomethylation was correlated (Table 5 and Table 6) with regulation of miRNA target genes, which had a reciprocal relationship with miRNA expression; genes with miRNA-targeted-gene hypomethylated promoters were downregulated during cell senescence15,16 and upregulated during differentiation (Table 8). I also found that genes with miRNA-targeted-gene hypomethylated promoters were specific targets of miR-548 miRNAs (Table 9 and Table 10). Regulation of target genes by miR-548 miRNAs, which were typically expressed at low levels are likely the result of miRNA-targeted-gene promoter hypomethylation. However, all findings here were correlative and thus lacked the consideration of biological background mechanism that causes the observed correlation.
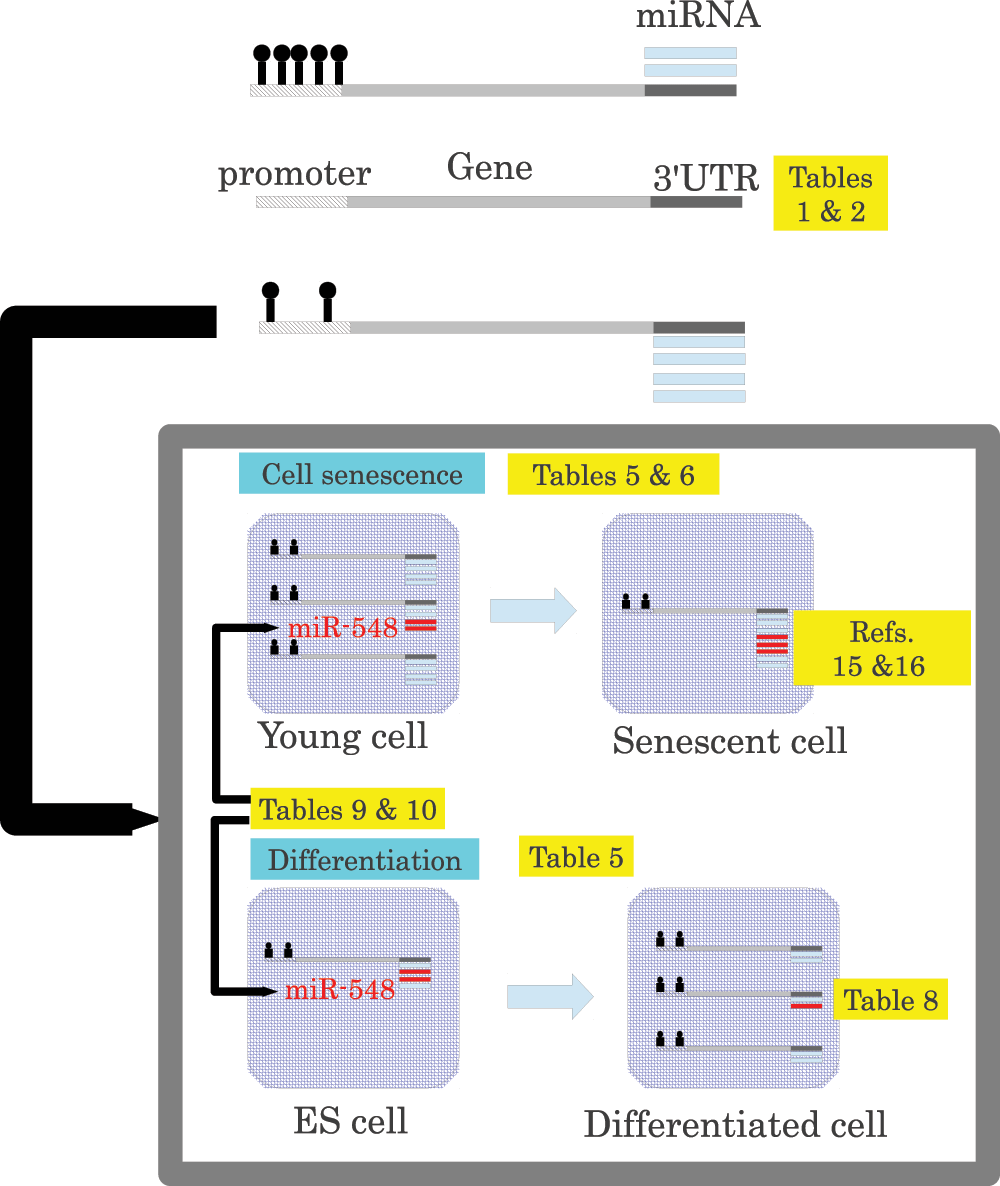
Figure 1. Schematic representing the principal findings of the present study together, as well as those reported previously.
Promoters are methylated dependent upon miRNAs that target each gene (Table 1 and Table 2). Genes with miRNA-targeted-gene hypomethylated promoters are downregulated during cell senescence and upregulated during cellular differentiation (Table 5 and Table 6), whereas miRNAs that target genes with hypomethylated promoters were upregulated during cell senescence15,16 and downregulated during differentiation (Table 8). miR-548 miRNAs, which are expressed at low levels, were suggested to regulate target genes with the assistance of miRNA-targeted-gene promoter hypomethylation (Table 9 and Table 10).
List of abbreviations
TSS: transcription start sites
GEO: gene expression omnibus
bp: basepair
miRNA: microRNA
ps-miRNA: primate-specific miRNAs
TE: transposable elements
NGS: next generation sequencing
RISC: RNA-induced silencing complex
Comments on this article Comments (0)