Introduction
Concept Profile Analysis (CPA) has proven a powerful tool for interpreting and prioritizing results of bioinformatics analysis, and for linking data sets based on the best “educated guess” when precise links are not available. The technology uses the vector space model to relate concepts (such as genes and biological processes) mined from the literature to each other. Vectors can be compared efficiently and transparently1, and the model yields a measure of the strength of the relationship between concepts. We call these vectors “concept profiles”. The CPA algorithms have for example been successfully applied to compare microarray studies2, for predicting proteins putatively associated with muscular dystrophy pathways3, and for associating chemical structures with gene expression data4.
The standalone application Anni10 supports a number of standard CPA operations. For example, to perform pathway analysis for a gene expression experiment, a user first provides a list of gene database identifiers for the most significantly expressed genes. Anni uses these identifiers to query the concept profile database for the corresponding concept profiles, and subsequently constructs a “concept set” of these profiles. To match the list of genes with pathways, the user performs the operation “match concept sets” for the gene concept set with a predefined concept set of the category “Gene Ontology (GO) biological process”. Note that we refer here to GO concept profiles. The concept profile matching scores between the two concept sets are calculated by Anni, resulting in a ranked list of GO biological processes for the gene list. Finally, literature evidence in the form of documents containing co-mentions of the gene and biological processes can be retrieved by Anni from a supporting documents database, or from documents providing enough statistical evidence to support the gene-biological process associations without actually mentioning the gene and the biological process together in an abstract.
Here we present a new suite of Web Service (WS) operations that allows bioinformaticians to design and execute their own CPA workflow outside the Anni Web tool, possibly as part of a larger bioinformatics analysis. The WS was designed according to the outcome of an Anni usage analysis, where the common user and machine operations were identified.
Technical specifications
We implemented the CPA WS using Java, Model-View-Controller (MVC) Spring framework, and Apache Tomcat following the Java API for XML WS (JAX-WS) specifications. We compiled the Anni Java code for the different operations into separate libraries, for which wrappers were written in Java. Spring MVC was used as a WS interface to remote applications. The WS was implemented according to the JAX-WS standard, enabling an auto-generated WSDL specification and use of Java Annotations to specify operations. Apache Tomcat was used for deployment. The CPA WS uses a database of indexed PubMed records. The thesaurus behind the Anni Web application was converted to Simple Knowledge Organization System (SKOS), and the SKOS concept IDs were implemented as resolvable Unique Resource Identifiers leading to a Virtuoso Universal Server triple store.
User and machine operations as Taverna workflows
As an example on how to work with the CPA WS we implemented several workflows in the workflow management system Taverna workbench v 2.45 following the best practices for workflow design6. The whole suit of CPA workflows consists of 11 workflows collected in a myExperiment pack [http://www.myexperiment.org/packs/368]. These workflows are of two different types: 1) nine workflows calling one WS operation, and 2) two pipelines of nested workflows calling more than one WS operation. The workflows of type 1 are the building blocks to make pipelines of type 2, and were implemented with re-usability in mind.
Here we describe the workflow “Match concept profiles with predefined set” (Figure 1) in order to illustrate the design and use of the WS and workflows. The workflow invokes the WS operation “getSimilarConceptProfilesPredefined”. The operation takes three input parameters, which can be accessed using the XML splitter function in Taverna. The user specifies the concept(s) to be matched (“Query concept IDs”), the concept set to match against (“Match concept set”), and a cutoff number of matched concepts to return (“Cutoff”).
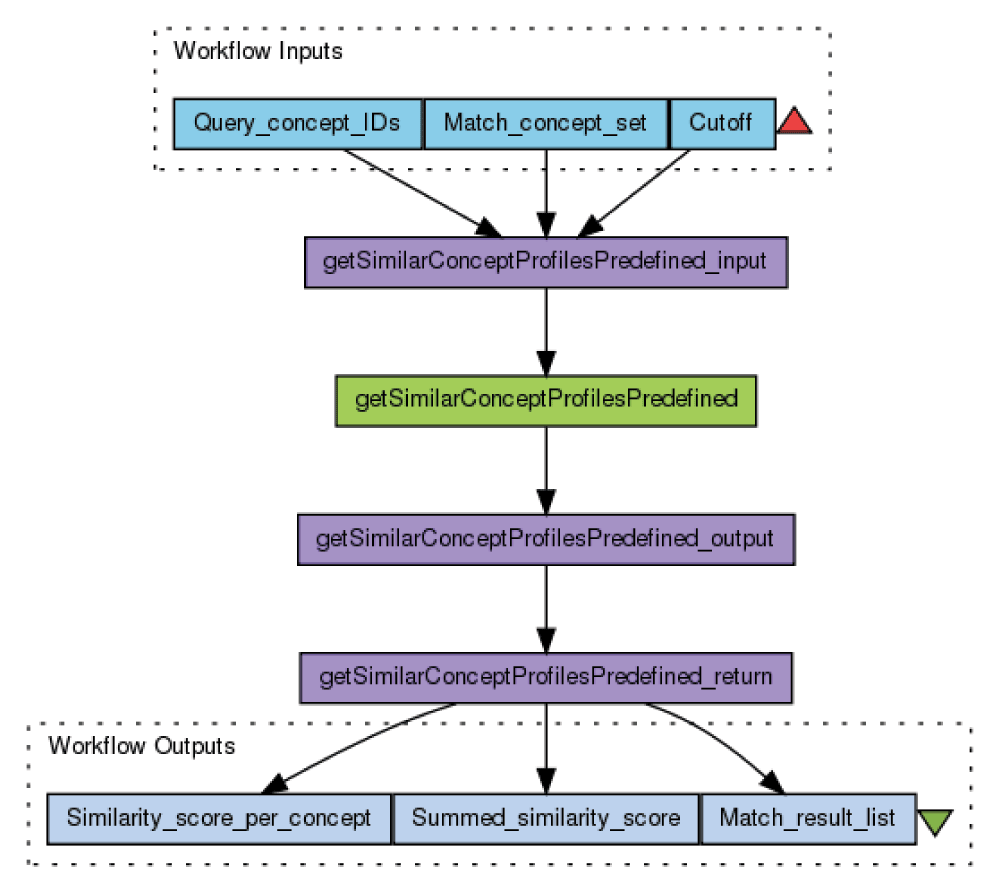
Figure 1. Taverna workflow for matching concept(s) with a predefined set of concept profiles.
Blue boxes represent the workflow inputs and outputs, green box the WS invocation, and purple boxes the XML splitters for the inputs and outputs of the WS operation. The workflow is available at http://www.myexperiment.org/workflows/3396.
Opening the “Run workflow” window in Taverna will result in showing the structured annotations for the whole workflow and the input parameters, as well as the example values (Figure 2). WS functional annotations can be accessed via the “Details” tab in Taverna (Figure 3). When the workflow is run, it will produce a ranked list of concepts associated to the query concept(s), and their similarity scores.
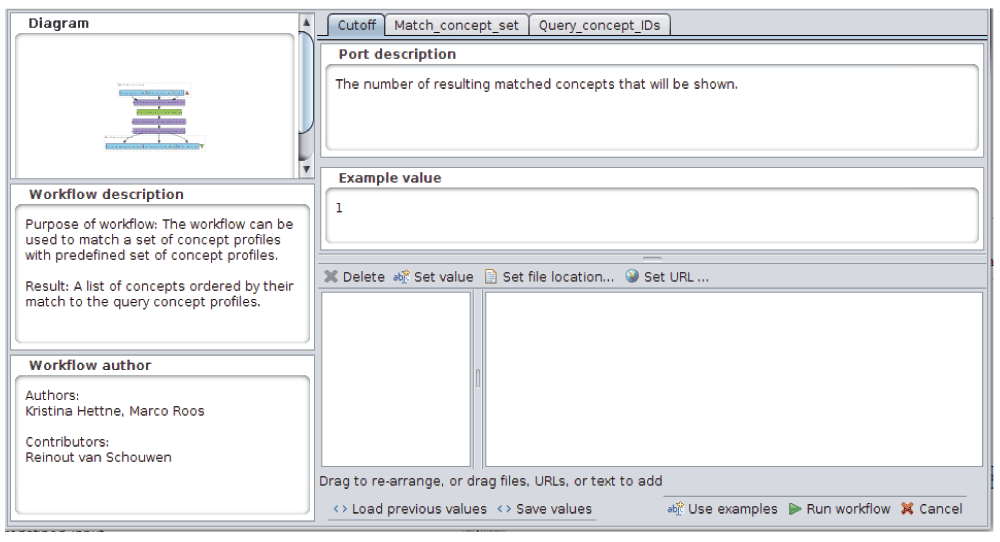
Figure 2. Taverna run window.
Detailed, structured descriptions for the whole workflow and its input parameters, with example values are shown in the window.
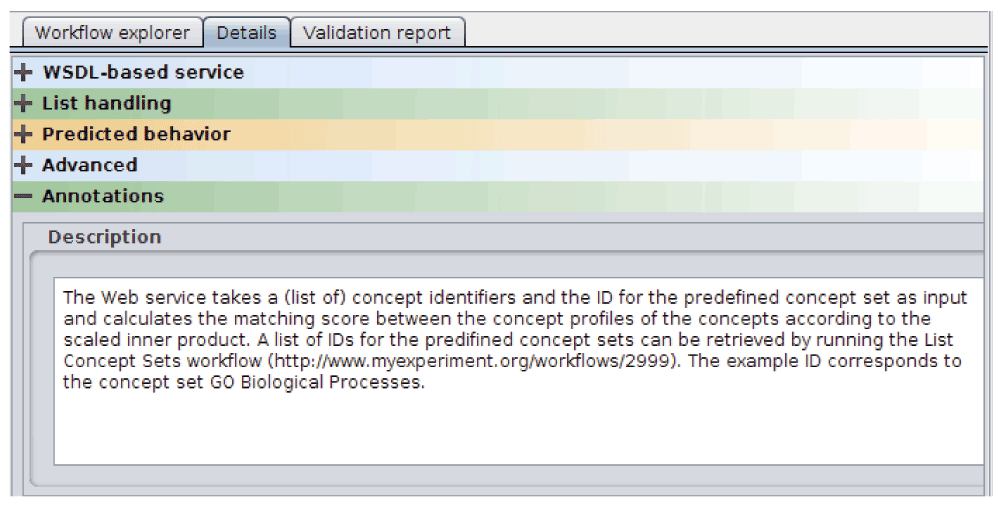
Figure 3. Taverna details window.
A detailed description of the function of the WS operation is shown in the window.
The above described workflow executes the core functionality of concept profile matching. The other WS operations implement functionality such as explaining the association found (by listing the common concepts contributing most to the score) and showing the literature evidence (by retrieving the links to the abstracts in PubMed). Workflows implementing these WS can be coupled to the “Match concept profiles with predefined set” workflow to form a pipeline of nested workflows. Examples of such pipelines are the “GWAS to biomedical concept” nested workflow, which performs Single Nucleotide Polymorphism annotation (SNP), and the “Annotate gene list with top ranking concepts” nested workflow for gene annotation (Figure 4).
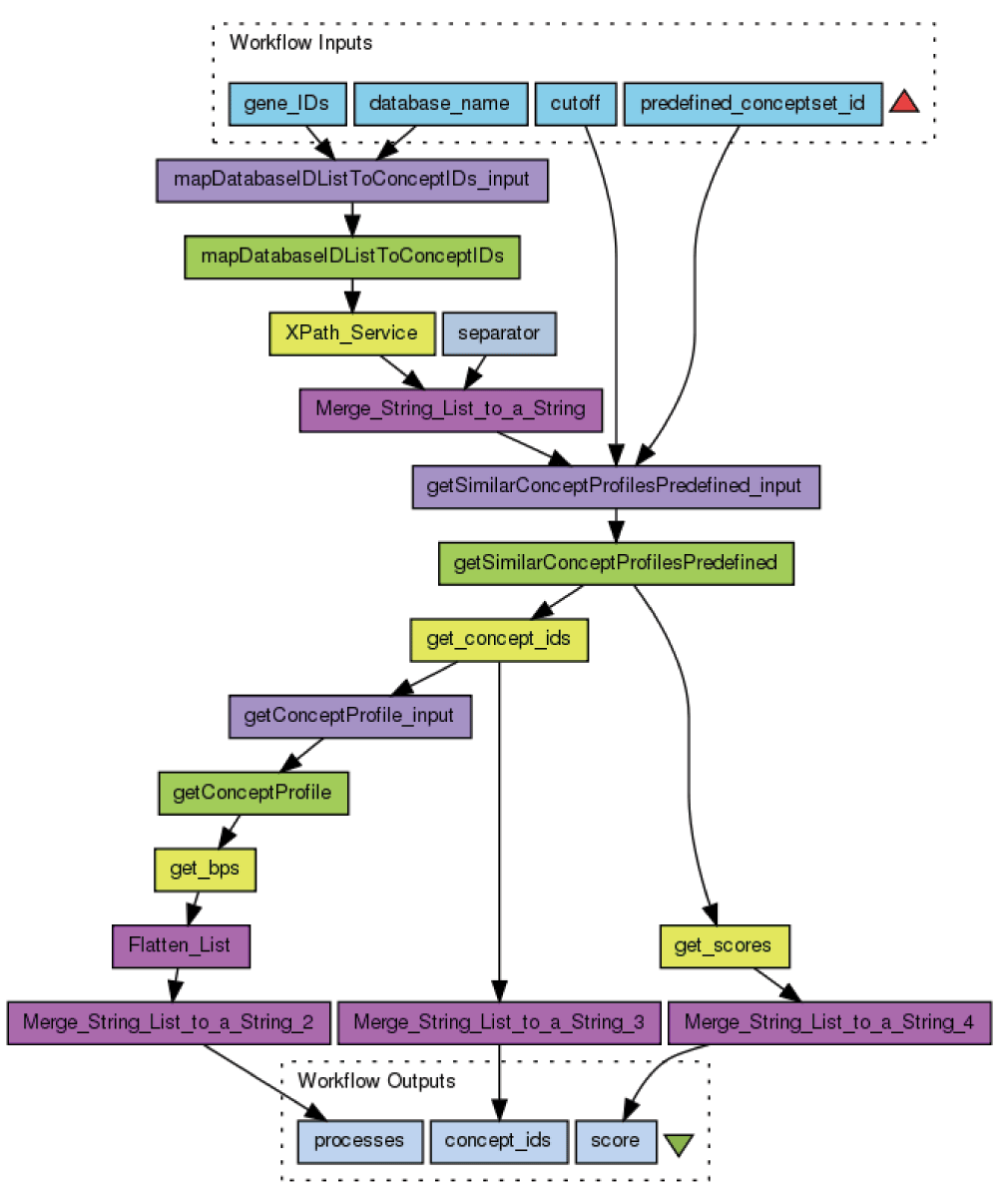
Figure 4. Taverna nested workflow for gene annotation.
Blue boxes represent input and output parameters, purple boxes the local Taverna worker services, yellow boxes the Xpath services for fast XML parsing, and grey boxes the constant values. The workflow is available at http://www.myexperiment.org/workflows/3921.
Comments on this article Comments (0)