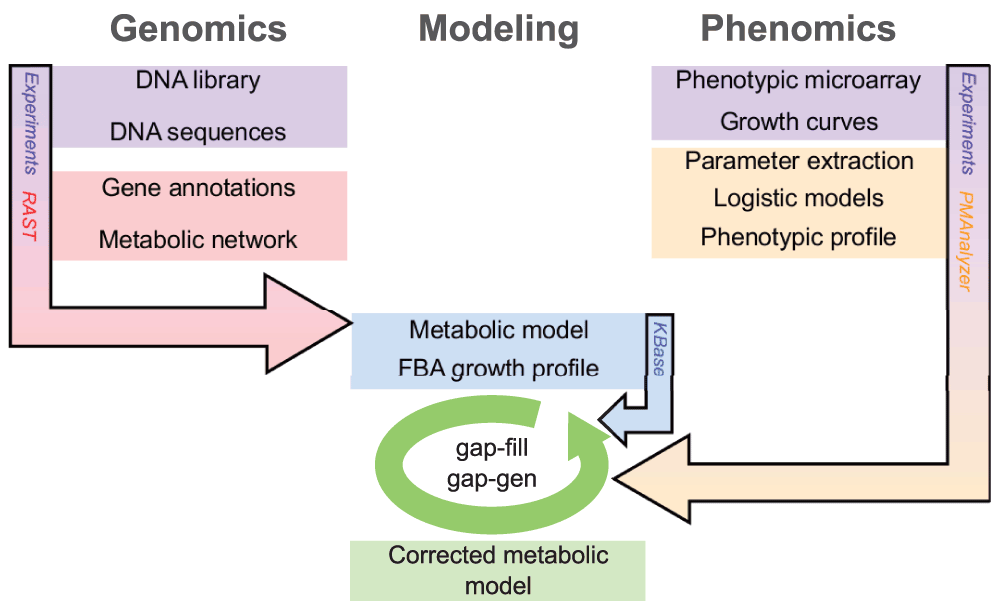
The article by Cuevas and colleagues represents an important attempt to streamline the assessment of phenotypic growth data in the context of genome scale metabolic models. The approach incorporates a non-proprietary phenotyping assay that should be accessible to a wide variety of research groups. The data are then coupled to a developing genome analysis and modeling environment,
KBase and associated systems (
the SEED and
RAST) to enable evaluation and iterative refinement of metabolic models. The primary contribution of the work is to provide a methodological path for acquisition and reasonably quick integration of these data with genomic information.
Below, I provide a series of questions, concerns and suggestions that I have for the each section of the manuscript as written.
IntroductionThis section is generally well written, though I think the context for this type of analysis could be better described... that is, beyond mentioning simply KBase and RAST. Perhaps providing rationale for potential advantages of using this combination of systems rather than other options would be useful for the reader.
In the final paragraph, the term "ground truth" is used to describe the phenotype data with respect to the metabolic modeling that will be performed. Foreshadowing some comments later, what is the evidence that the phenotype data are actually correct for all conditions tested?
MethodsFirst, thank you for the detailed description of the methods. These are clearly written overall. Some detailed comments/suggestions.
Describing growth of the initial cells and cultures from glycerol stocks, please define the shaking parameter (rpm) and define "agitation". These parameters can be critical factors for reproduction of experiments and are often organism dependent.
Second full paragraph, you have defined the contents of 1X MOPS, but you also define it two paragraphs later in the context of the recipe for the basal media. This first instance could be removed and simply refer to the recipe later described.
Question: In the plate format for growing the cells, you indicate that plates are sealed with a PCR grade plate film. What does this do to the aerobic/anaerobic state of each well? Is there any opportunity for gas exchange during incubation? Also, is there any shaking going on during incubation on the plate reader? It might be worth mentioning about caveats of usage of carbon/nitrogen sources being limited to these conditions, which aren't exactly known.
Also, the modeling could be impacted by the aerobic/anaerobic status of the environment. Was modeling performed under both conditions? Would this impact the accuracy of the modeling results?I note the storage condition was at room temperature. Are all of the substrates stable at room temperature? How long would each stock be stored prior to use for replicates?
In the "Sequencing and metabolic reconstruction..." section:
- The sentence that starts with "FBA was used to determine...". This is a confusing sentence. What is meant by this?
- The KBase workspace, Citrobacter_sedlakii_119, does not appear to exist in the current public release of KBase (as of March 25, 2015). I also am unable to find any FBA model objects searching for various forms of Citrobacter and sedlakii. There are not an public narratives that would match the series of commands that you describe as being run and as freely accessible. This needs to be corrected, likely by building a public narrative in the current system.
- Are the named commands for KBase still valid in the current production version of the system? It would be useful to include what apps and methods correspond to these commands. A public narrative in the current version of KBase would make this study replicable and easily transferred to other model systems.
The github page for the PMAnalyzer software is good... to the point, clear.
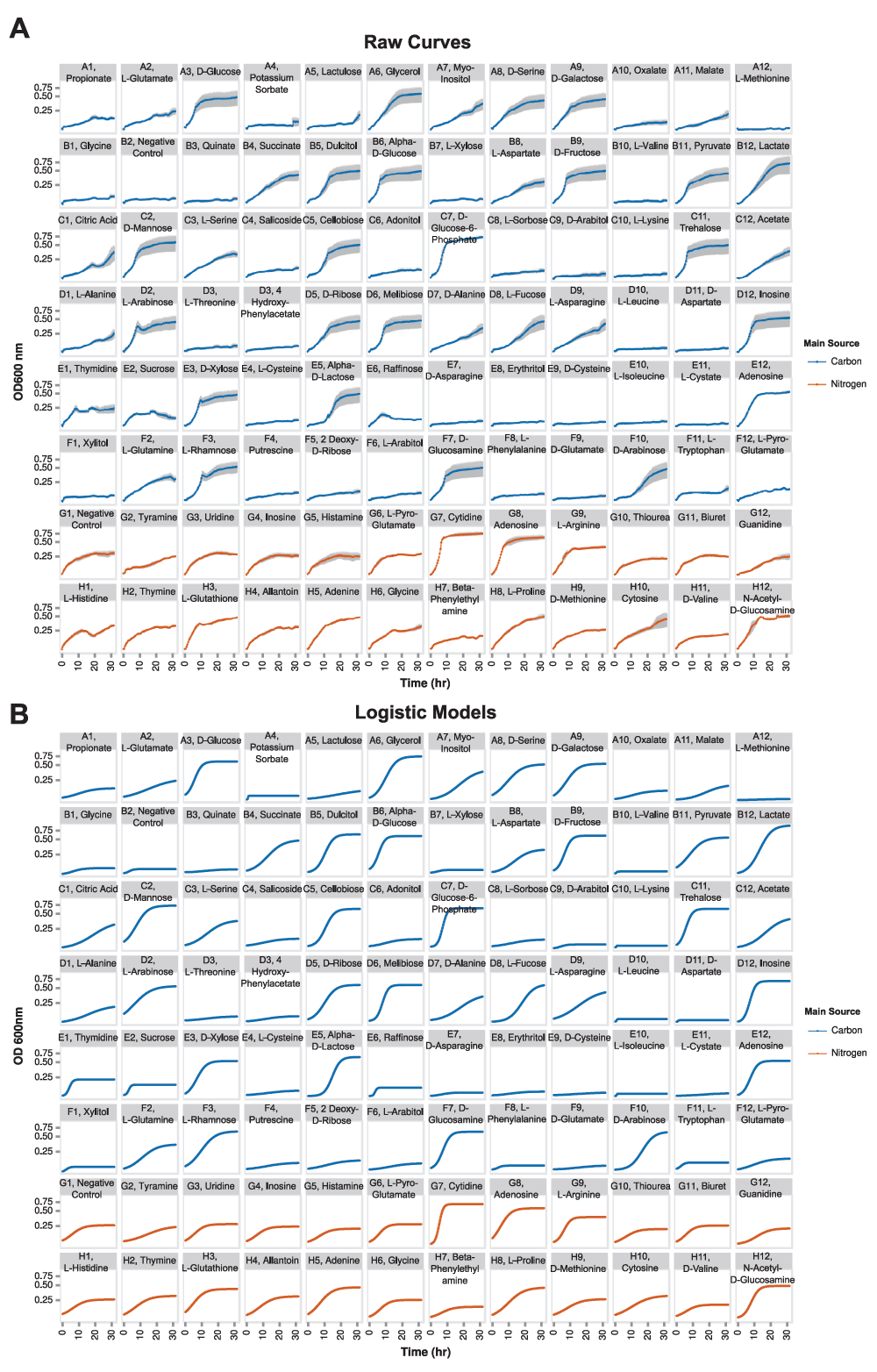
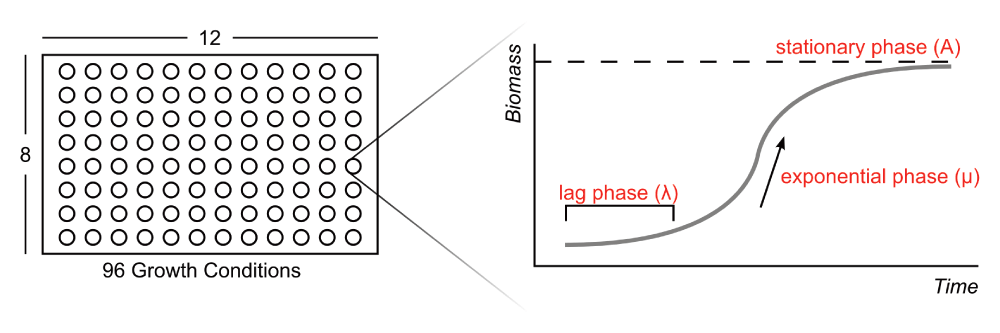
The explanation of the logistic model and absorbance data is also clear. In the description of the
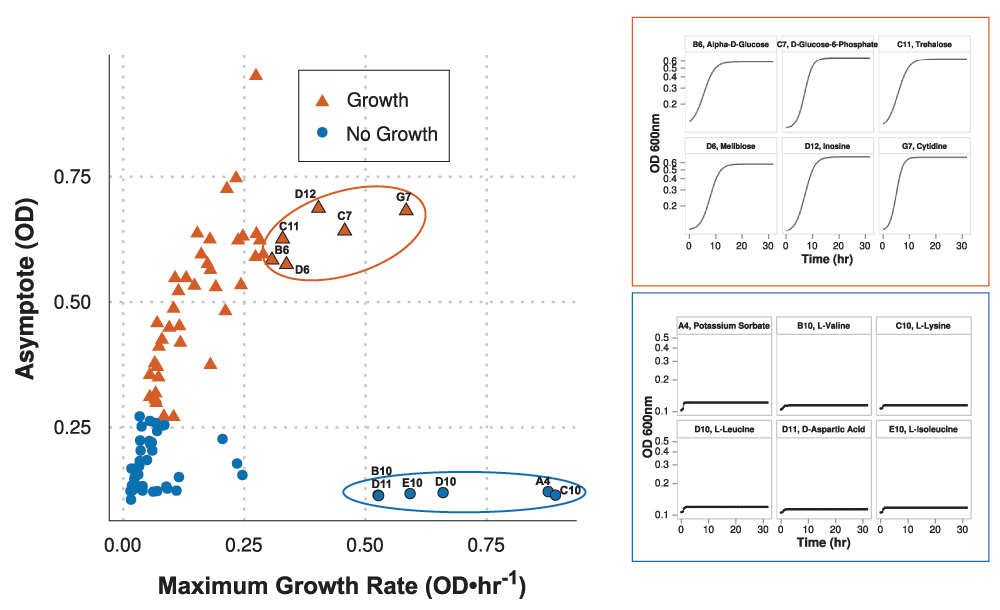
growth value, you end by stating that this is boiled down to a boolean growth/no growth status for each condition. I understand why this is done, given that the model reconciliation with growth phenotypes is occurring on a boolean level, but how much information is being lost by making this experimental design decision? The nature of the growth can be very important for understanding how the organism is behaving in an environment. The more immediate consequence of this decision is in the interpretation of False Negatives by the model (where the phenotype assay says "growth" and the model says "no growth").
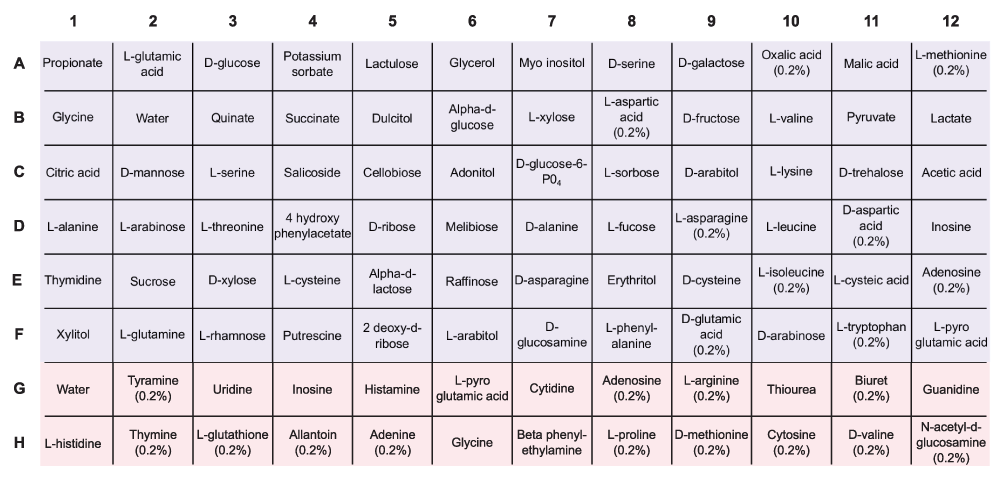
How many of the false negatives had growth values near the 0.5 cutoff? The allantoin example could be a case like this (growth = 0.529, from curve_logistic_parameters.csv). The growth curve asymptote appears to be near 0.25 (Fig. 2a). This is very similar to values that are considered "no growth" phenotypes. Does it make sense to have the model gap fill three reactions in this case? Table 1 might be made more complete by adding a column for the growth value for each condition rather than that sitting in the supplemental data files (alternatively, highlight in the text that these values are given in that file). Related to this, in Supplemental Figure 3, you could highlight the point that represents allantoin. It would also be useful to highlight the water, negative control in the Supp. Fig. 3.This brings me to questions about how confidence in the
growth value is determined (if at all). I see that the median value of the biological replicates is used to determine the y logistic and thus the
growth value. I also see that standard error is indicated in Fig. 2a graphs. However, this does not allow for statistical evaluation of the
growth.
Would it be better to calculate growth for each replicate independently and then determine an average growth value with error around these? Perhaps there is a better statistical approach. In any case, this comes back to being able to state some confidence in these values to aid interpretation of potentially borderline cases. Please define "sse" in the curve_logistic_parameters.csv file.
In the "RAST annotations" section, last paragraph. How does this fit in with the gap filling process for the model? Is the context information in close genomes actually used in the gap fill process, or is it a post hoc attribution of higher confidence to the gap fills that are included in the model?
ResultsThe statistics on the genome assembly are worse than I would expect to see. In particular, is the coverage based on alignment by blastn to
C. koserii a reasonable number? I can't quickly evaluate if this is typical of different
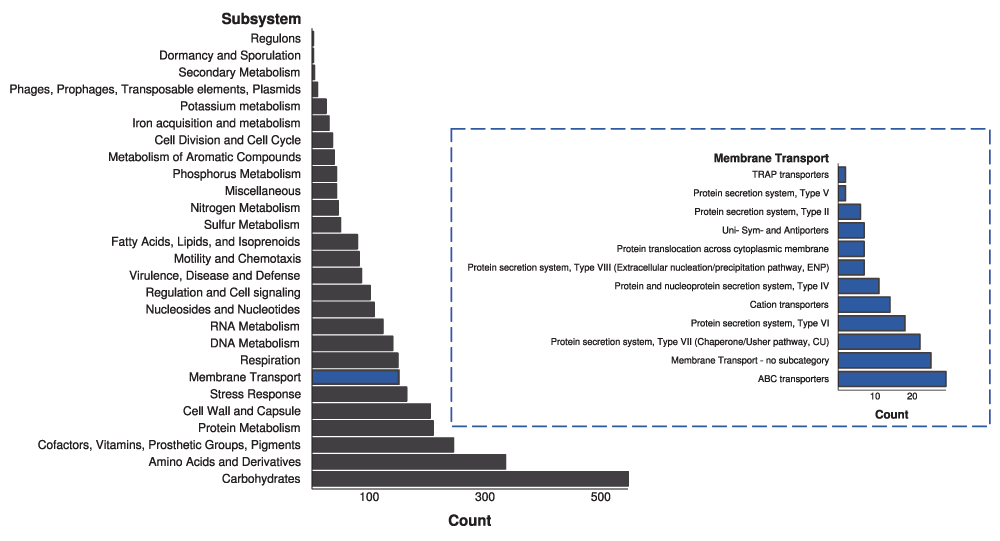
Citrobacter genomes. How does a low coverage (~70%) affect the outcome of presence/absence of genes in the annotation and subsequent modeling process. In reading the results, it appears that the majority of reactions in the network are identified, but it may be worth addressing this explicitly.
I note that you used manual inspection of growth curves just under the 0.5 cutoff... this is another area in which a statistical confidence in that value might help. If this is to become truly high throughput, manual inspection becomes untenable except in a few cases.
In the description of gap filling reactions for complex media, you mention that EC 2.2.1.7 was not found, but is likely due to a frameshift error. Was any follow up sequencing or PCR performed to confirm the error or presence/absence of the gene in
C. sedlakii? Or even just a blastx analysis of the region?
In general, when you are discussing the evaluation of annotations in RAST, figures (supplemental) of key genomic regions would help the reader to evaluate the statements being made.In the paragraph beginning with, "Using the base model, the 90 well simulation resulted in...", you have a sentence that starts with, "Note:". This is a confusing sentence and structure. What are you trying to point out here? What reactions are being referred to?
What percentage of gap filled reactions are transport reactions? Stating this clearly would improve clarity.
The last statement in the Results section focuses on false positive conditions.
Do you have any thoughts as to why these are coming up as FP? There is no follow up in the discussion about this. Are they central in a network of reactions, are they dual use reactions, etc.? Discussion RAST annotations and gap-filled reactions section:
This section would also benefit from a supplemental figure that serves as an example of what is being discussed (also mentioned above).
What is the connection between KBase, RAST and SEED? How does updating in one affect the others? This question gets at an assumption in the text that the relationships among systems are known to the reader. The text could be clarified, or key references added.
FBA false positives section:
Please expand to include more specific discussion of the 6 FP reactions identified at the end of the Results section. What are the
growth values for these? Are any of them borderline?
Last paragraph:
It would be good to quantify what “several” means with respect to the number of metabolic pathways being targeted.
“…available in a day of using RAST and KBase.” This sentence implies that sequencing, annotation, and model reconstruction can happen in a single day. This should refer only to the use of sequence data. Also, there in no mention of the phenotype data here in this context. I think it would be better to highlight that the system allows the user to produce a reasonably robust metabolic model quickly, giving more opportunity for in depth analysis of discrepancies and manual curation of the model given the phenotypic data.
What is the link to the web service for the PMAnalyzer? Points to Address I’ve bolded several items in the above format for this review that I would consider to be major points to address and would make the manuscript stronger. Given that this is a methods paper, it is imperative that others can reproduce the work and/or employ the approach in other organism systems. Please update methods as requested above, paying particular attention to the KBase functionality and workflow.
Non-financial competing interests include: I have worked as a collaborator with several of the authors on projects related to the SEED, RAST and KBase. I have not worked directly with the lead author.
Financial competing interests: None to declare.
Comments on this article Comments (0)