Keywords
high-throughput model reconciliation
high-throughput model reconciliation
Version 2 contains changes in the text and figures that clarify the ambiguities recognized by the referees. Figures 1, 2, and 3 have been revised. Supplementary Figure 4 has been added based on suggestions by the referees. We have also made available the metabolic models in SBML format. We thank the referees for their time and comments.
See the authors' detailed response to the review by Matthew A. Oberhardt
See the authors' detailed response to the review by Aaron Best
Recent advancements in genomic sequencing provide high quality, deep-coverage DNA sequences for tens of thousands of bacterial genomes. To manage this breadth of data, online tools such as RAST (http://rast.nmpdr.org/)1 leverage the SEED database2 by using homology and genomic context to determine gene functions encoded in the DNA sequences. This automated annotation service additionally generates a raw metabolic reconstruction of the genome for use in in silico experiments. Genome-scale metabolism analyses use these reconstructions as input into data environments such as KBase, the Department of Energy Systems Knowledgebase (http://kbase.us). Several hypotheses can be tested simultaneously, e.g., protein function identification, biological behavior simulations, and metabolic network comparisons3.
Metabolic models are defined by the chemical reactions that characterize the vast metabolic network of an organism. Flux-balance analysis (FBA) uses these chemical reactions to provide understanding of the physiological capacity of the cell4. Mathematically, the stoichiometry of metabolic networks is represented by a two-dimensional numerical matrix, in which the values are the stoichiometric coefficients of the reactants and products. Each row and column in the matrix is associated with a metabolite and a metabolic reaction, respectively. For one stoichiometric reaction, the products of the reaction are given positive integers, the reactants are given negative integers, and non-associated metabolites are given zeros. Through a constraint-based approach, the FBA algorithm uses linear programming techniques to solve this system of stoichiometric coefficients, optimizing for biomass production or another objective function4–6.
The amount of published metabolic reconstructions for prokaryotic and eukaryotic organisms has increased over the past decade3,7. Through the increased use of next-generation sequencing and automated annotation software, metabolic models for new organisms are arising and older models are continuously being reconciled. However, a drawback to RAST and other gene annotation algorithms is the dependency on previous functional annotations. The breadth and quality of annotated functions vary among and across bacterial species, which is not accumulating as quickly as new sequences. Automatic generation of metabolic models is limited by our knowledge of cellular metabolism and biochemistry. In addition, an existing problem with gene databases is the inconsistent nomenclature used to name and define the function of a gene. Separate databases hold slightly different annotations for the same gene, which propagates into downstream tools, leading to a loss of information in analyses as mis-annotations cause reactions to be missing in models built during the initial reconstruction. To bridge the gap between quality genome annotations and accurate metabolic models novel methods are needed to supplement the reconciliation process.
Multi-phenotype Assay Plates (MAPs) provide a system to quantitatively monitor microbial growth while qualitatively deducing the metabolic capabilities of a microbe across a range of conditions. The MAPs technology uses optical density to measure biomass production by substrate utilization of a clonal microbial population. MAPs and similar technologies, such as Biolog’s Phenotype MicroArrays, have been an advantageous tool in past phenotypic studies3,8–20,29. Although the Biolog system similarly measures substrate utilization, it does so as a response of cell respiration (i.e., reduction of a tetrazolium dye) and not specifically biomass production. Using MAPs, we can measure bacterial growth in defined conditions to aid in the validation of microbial genome annotation software.
In this study, a metabolic model of Citrobacter sedlakii is built using a workflow combining experimental data and computational analysis (Figure 1). The genome of C. sedlakii was sequenced, annotated, and a metabolic reconstruction was subsequently generated using RAST and the KBase platform. Growth of C. sedlakii was measured in 96 different growth conditions and the resulting data was introduced into a novel computational pipeline, PMAnalyzer (https://vdm.sdsu.edu/pmanalyzer). The PMAnalyzer automatically parameterizes raw growth data and fits a logistic model of bacterial growth for each experimental condition. Observed phenotypes from the MAP experiments were used to ground truth the genome-scale metabolic model by running FBA simulations on the KBase platform, which identified disparities in the metabolic reconstruction. Disparities were observed to have either been missed due to RAST mis-annotations or sequencing. Here, we introduce a high-throughput workflow to obtain large-scale metabolic reconstructions and reconciliations with observed growth phenotypes.
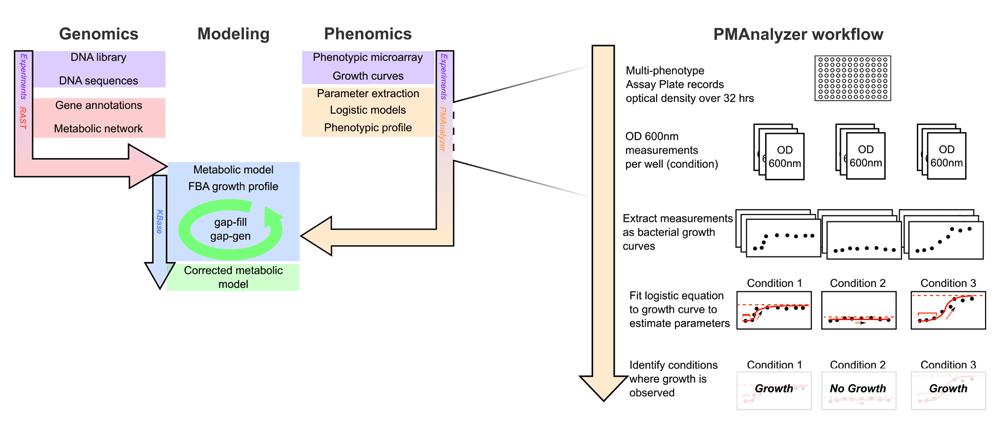
Analysis pipeline for generating and reconciling metabolic models. Initial metabolic models are built and reconciled in KBase from RAST genome annotations. Phenotypic profiles from the MAPs technology and the PMAnalyzer are incorporated into the KBase FBA-reconciliation loop to optimize the model. On the right are details of the PMAnalyzer workflow portraying the production of growth profiles from the MAPs.
The C. sedlakii isolate (ATCC 51115, CDC 4696-86) was provided by Dr. Marlene DeMers in the Department of Biology at San Diego State University. A glycerol frozen stock of the sample was plated on trypticase soy agar (Becton, Dickenson and Company) and incubated at 37°C for 24 hrs. A single colony was inoculated into 3 mL of trypticase soy broth (Becton, Dickenson and Company) and incubated, with shaking, at 37°C for 24 hrs. 500 µL of the overnight culture was mixed with 500 µL of 30% (weight/volume) filter sterilized glycerol and transferred to a cryogenic vial (Fisher Scientific) for storage at -80°C.
C. sedlakii was grown overnight at 37°C for 24 hrs on 50% Luria-Bertani (LB) agar (Fisher Scientific) from a frozen glycerol stock. Three independent colonies, biological triplicates, were inoculated into 3 mL of a modified 3-morpholinopropane-1-sulfonate (MOPS) broth21 (1X MOPS (40 mM MOPS + 10 mM Tricine), 0.4% glycerol, 9.5 mM NH4Cl, 0.25 mM NaSO4, 1.0 mM MgSO4, 1.32 mM K2HPO4, 10 mM KCl, 0.5 μM CaCl2, 5 mM NaCl, and 6 μM FeCl3) and incubated for 24 hrs at 37˚C with agitation (250 rpm). Overnight cultures were centrifuged using an Eppendorf Centrifuge 5418R at maximum speed (14,000 rpm) to pellet cells and washed with 500 μL of 10 mM Tris/10 mM MgSO4 buffer, twice. Cells were re-suspended in 1 mL of 10 mM Tris/10 mM MgSO4 buffer and optical density at 600 nm (OD600) was measured using a Beckman Coulter DU 640 spectrophotometer. All suspensions were concentrated to achieve a final optical density of approximately OD600 = 0.1 after a fifteen fold dilution.
10 µL of concentrated cells was transferred into each well of a sterile 96 well, micro-titer plate (Grenier Biosciences), which contained 60 µL of sterile water, 50 µL of 3X MOPS basal media, and 30 µL of 5X substrate (Supplementary Figure 1). Each plate was sealed with PCR grade plate film (Sigma SealPlate® film) with gas exchange still possible, and incubated on a Molecular Devices Analyst GT multi-plate plate reader (Molecular Devices, LLC.). Plate reader was programmed to incubate MAPs at 37°C and measure OD600 every 30 min with shaking before each read, for a total of 32 hrs. Absorbance data was saved, extracted as a text file, and uploaded to the project website for data storage (http://vdm.sdsu.edu/).
MOPS basal media is derived from the culture media provided by Neidhardt et al.21, and contains 1X MOPS (40 mM MOPS + 10 mM Tricine), 0.4% glycerol*, 9.5 mM NH4Cl*, 0.25 mM NaSO4*, 1.0 mM MgSO4*, 1.32 mM K2HPO4*, 10 mM KCl, 0.5 μM CaCl2, 5 mM NaCl, 6 μM FeCl3. Media was prepared with sterile Milli-Q water (Milli-Q Integral Water Purification Systems, EMD Millipore) and subsequently filter sterilized using 0.22 µm Sterivex filter unit (Millipore, Inc). (*These compounds are not included depending on the basal media. For example, 0.4% glycerol is not used in the carbon basal media, while 1.0 mM MgSO4 is replaced by 1.0 mM MgCl2 in the sulfur basal media).
Nutrient substrates were prepared by dissolving 1.25% (w/v) of the solid compound in sterile Milli-Q water and filter sterilized with a 0.22 µm Sterivex filter unit (Millipore, Inc). Substrate stocks were stored at 5X concentrations at room temperature in sterile conical tubes. Supplementary Figure 1 contains a detailed mapping of the substrates used in the MAPs.
As part of a DNA sequencing class at San Diego State University26 the C. sedlakii 119 genome was sequenced using 454 pyrosequencing with the GS Junior platform and assembled with Newbler version 2.7. RAST (http://rast.nmpdr.org/) was used for subsystem annotations and metabolic reconstructions1. Annotations were imported into the KBase environment where the metabolic model was viewed, manipulated, and used in flux-balance analysis (FBA) simulations. FBA was used to determine if the model bacteria is successful in ascertaining growth in specific conditions equivalent to the MAPs. The KBase command kbfba-importfbamodel was used to import the annotations into the Citrobacter_sedlakii_119 workspace and was named C.sedlakii_nogapfill. The model is also available in the supplementary files in SBML format in Dataset 1. The Citrobacter_sedkakii_119 workspace and its objects are freely accessible to anyone. Using the KBase command kbfba-gapfill, initial gap-filling was performed on the model while specifying Luria-Bertani (LB) as the growth condition (the default ArgonneLBMedia formulation was used). The reconciled model was named C.sedlakii_ArgonneLB_gapfill and is provided in the workspace. This gap-filled model is available in the supplementary material in SBML files in Dataset 1. The LB gap-filled model created a representative model that fulfills the general requirements needed to utilize a rich media source for growth.
The high-throughput analysis pipeline described below, and in Figure 1, was executed in a Linux command-line environment and was developed using several programming languages, including bash, Perl version 5.16 (http://www.perl.org/), and Python version 3.4.1 (http://www.python.org/). Perl scripts were written to parse and format the MAP raw data files into the tab-delimited intermediate files. The primary analysis script (Python) used these intermediate files for modeling the growth curves. For ease of execution, a single bash program was created as a wrapper script that executes the parsing and analysis scripts as a cohesive, automated pipeline. Command-line arguments or a configuration file was used for user input and settings. All scripts are freely accessible from a Git repository at https://github.com/dacuevas/PMAnalyzer. The online implementation can be found at https://vdm.sdsu.edu/pmanalyzer.
Phenotypic responses were recorded by measuring the optical density at 600 nm (OD600) over time, which quantitatively represents the bacterial biomass concentration at each time point. The OD600 values are plotted to form the sigmoidal shape characteristic of bacterial growth curves. This characteristic curve, highlighted by Monod22 and modeled by Zwietering et al.23, consists of three phases: lag, exponential, and stationary phases. Zwietering et al.23 interprets these phases as parameters required to model growth, using his logistic equation
where y0 (OD600) is the starting optical density, λ (hr) is the lag phase, μ (OD600•hr-1) is the maximum growth rate during the exponential phase, A (OD600) is the asymptote of the growth curve representing the carrying capacity of the population, and t (hr) is time. Supplementary Figure 2 provides a visual representation of a classical growth curve.
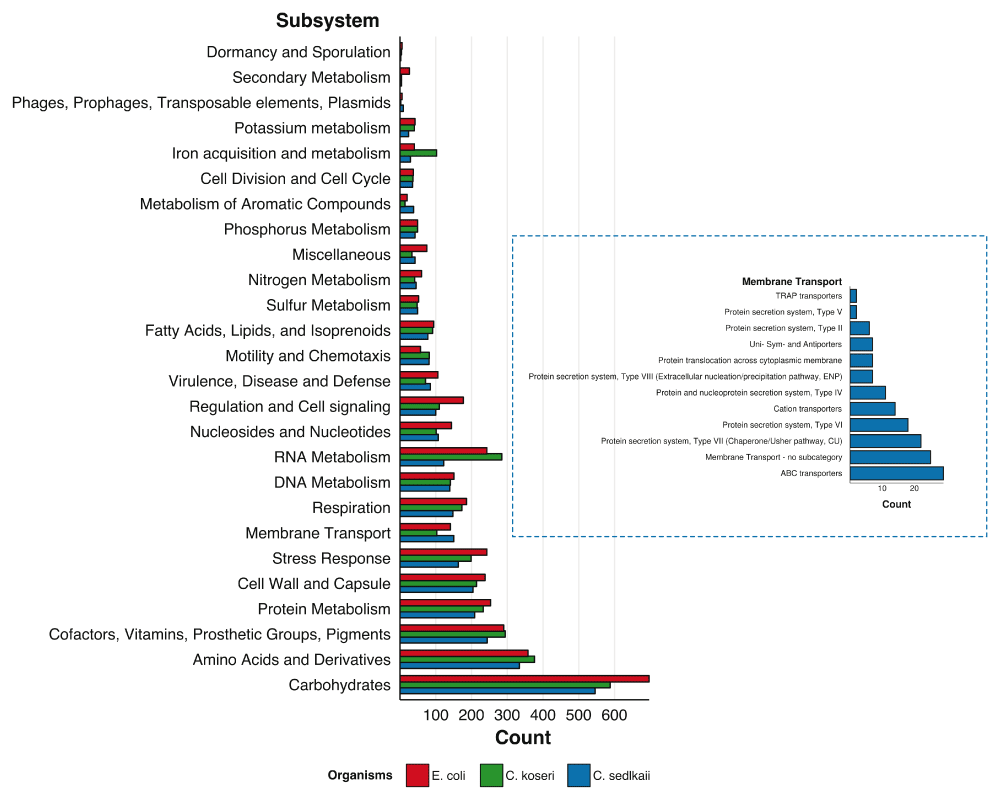
Number of subsystems annotated by RAST for each subsystem group for Citrobacter sedlakii and two phylogenetic neighbors Citrobacter koseri and Escherichia coli. Membrane transport subsystems identified in C. sedlakii are highlighted in separate plot.
To parameterize the growth curves median values of the replicates were used. Python’s NumPy module version 1.8.1 and SciPy module version 0.14.024 provides several functions for optimizing nonlinear, multivariate functions. In this case, the minimize function was used in order to denote bounds and constraints on each parameter. The default Broyden, Fletcher, Goldfarb, and Shanno (BFGS) algorithm25 was used to minimize the sum of squared error between the logistic model from (1) and the raw data. As input, the algorithm requires estimations for each growth curve phase. The estimation for the asymptote was defined as the largest OD600 reading from three consecutive time points (2) and the maximum growth rate was defined as the largest change in OD600 over a 1.5 hrs window (3). The estimated lag time was set at 0.5 hr.
The result from (Equation 2) was also used as the upper bound for the minimization function. Lag time and maximum growth rate were not given an upper bound. Lower bounds for the asymptote, maximum growth rate, and lag time were 0.01, 0, and 0, respectively.
Each well in the MAPs has a varying level of growth, including different lag times, maximum growth rates, and asymptotes. A single value that represents the overall level of bacterial growth per well was generated by adapting the logistic model with the asymptote:
Here, is the value from (1) at time i, and n is the number of data values used, which is the number of OD600 measurements recorded during the experimental run. For the C. sedlakii MAP, n equals 64. The asymptote factor A, rather than the maximum growth rate, contributes to defining growth (i.e., growth levels from wells that achieve a higher biomass yield separate from those growth levels of wells that exhibit less growth, Supplementary Figure 3). In certain instances, growth curve models were fitted with a high maximum growth rate but did not display growth (e.g., potassium sorbate, L-valine, L-lysine, L-leucine, D-aspartic acid, and L-isoleucine). Ultimately, (4) was implemented to distill each growth curve into a single boolean variable of growth (≥ 0.5) or no growth (< 0.5).
The kbfba-simpheno function in KBase executes multiple flux-balance analysis (FBA) processes in parallel. To perform this, a text file listing information on which media condition to use as the input media in each process is required. Information regarding the PMAnalyzer result, i.e., growth or no growth, for each media condition tested on the MAPs are also listed in the text file. Digital representations of rich LB media and 90 different media compositions used in the MAPs were generated as media data objects in KBase. Each media object represents a specific condition used in the MAPs. kbfba-simpheno performs a separate FBA on each media object and compares the result to the MAPs result listed in the information text file. KBase FBA results are labeled as: Correct Positive assertions (FBA and MAPs both display growth), Correct Negative assertions (FBA and MAPs both display no growth), False Positive assertions (FBA asserts growth, MAPs display no growth), and False Negative assertions (FBA asserts no growth, MAPs display growth). Gap-fillings were attempted for conditions associated with false negative assertions and gap-generations were attempted for instances of false positive assertions. As stated previously, FBA and reconciliation was performed on the LB condition first in order to identify and integrate missing reactions required for growth on general, rich media. Thereafter, using the base model capable of asserting growth on LB, FBA and reconciliation was performed on the minimal media conditions with false negative assertions to target missing reactions in specific metabolic pathways. The minimal set of reactions determined by gap-filling was integrated into the model using the KBase function kbfba-integratesolution. To verify that the integration of new reactions produces additional correct positives and correct negatives the multi-FBA simulation was re-executed. Subsequently after gap-filling and multi-FBA simulation, some growth conditions resulted as false positives. Conditions where the model correctly asserted no growth prior to gap-filling had changed to incorrectly asserting growth. The KBase function fba-gapgen was executed on those conditions to identify a set of reactions to alter or remove from the model that will resolve the false positive into a correct negative result. This algorithm attempts this without altering the outcome of a correct positive result condition, thus ensuring critical reactions for the model to grow are not removed.
The gap-filling process provides biochemical processes that are missing from the initial model, and understanding why these reactions were missing during the initial reconstruction is important to investigate. The genomic databases available are numerous and nomenclature can differ between these resources. Reaction names for functions of genes may vary between databases, thus, resulting in a disconnect between gene annotations and the biochemical reactions those genes are involved in. When identifying functions for metabolic reconstructions, this unclear nomenclature prevents some reactions to appear in the initial metabolic model, thus producing a “mis-annotation”. To correct for this, following gap-filling, all missing reactions (excluding transporters and newly-modified bidirectional reactions) were cross-checked with the SEED database to find similarly named reactions. The new list consisted of a mapping of gap-filled reaction names to possible alternative names. This list of reactions was then referenced back to the C. sedlakii RAST annotations in order to determine if the reaction was identified by RAST as the alternative name but not included in the metabolic model. A successful match between an alternative name (from the cross-check list of gap-filled reactions) and a name from the original RAST annotations would mean the KBase system failed to include a reaction whose enzyme was identified during annotation, and therefore, should have been included in the initial metabolic model. When the search similar nomenclature did not resolve, a search for gap-filled reactions in closely related organisms was performed; i.e., Citrobacter koseri and E. coli K12. This consisted of Protein BLAST (blastp) searches of the reactions’ sequences from the SEED database against C. koseri and E. coli. Gap-filled reactions that are present in C. sedlakii’s closely related genomes were included in the C. sedlakii model with high confidence since closely related taxonomic groups contain common genetic material and function.
The genes encoding the missing reactions may be present in low quality DNA sequences or low coverage genomic regions. Following sequence assembly, these sequences are not present within the contigs, preventing RAST from annotating the proposed function. However, neighboring genes or protein complexes may be present and annotated, suggesting that the gene in question is there but was poorly sequenced. From within related organisms, the protein sequences of these complexes were identified and searched against the C. sedlakii genome. Finding matches for neighboring genes increases the confidence of including the reaction into the model. This method is also applicable to those genes that were not sequenced or that fall between assembled contigs.
C. sedlakii 119 was assembled into 320 contigs containing 4,604,104 nucleotides with an N50 of 28,039 bp. RAST annotated the genome as containing 4,035 protein encoding genes and 76 tRNA genes over 537 different subsystems (Figure 2). Hypothetical proteins consisted of 817 (~20%) of the protein coding sequences. Membrane transport features constituted 150 out of 3,031 subsystem features. RAST listed E. coli as a close neighbor and Citrobacter koseri as the closest Citrobacter neighbor. A BLASTn alignment (expected value of 10-4)27 against the C. koseri ATCC BAA-895 genome (NC_009792.1) resulted in a whole genome coverage of 69.8% (69.6% when including plasmids (NC_009793.1 and NC_009794.1). Figure 2 also displays subsystems for the C. koseri genome and the Escherichia coli K12 genomes, two close phylogenetic neighbors of C. sedlakii whose genomes are publicly available in the SEED database.
Growth on MAPs of each biological triplicate was followed for 32 hrs on separate days. After completion, the OD600 readings were processed through the PMAnalyzer pipeline (including data parsing, curve-fitting, and growth level analysis) in 8 seconds. Using a 0.5 growth level (4) cutoff and manual inspection of curves falling under the cutoff, 48 out of the 90 growth conditions – 35 carbon-based media and 13 nitrogen-based media – exhibited growth (Figure 3 and Table 1).
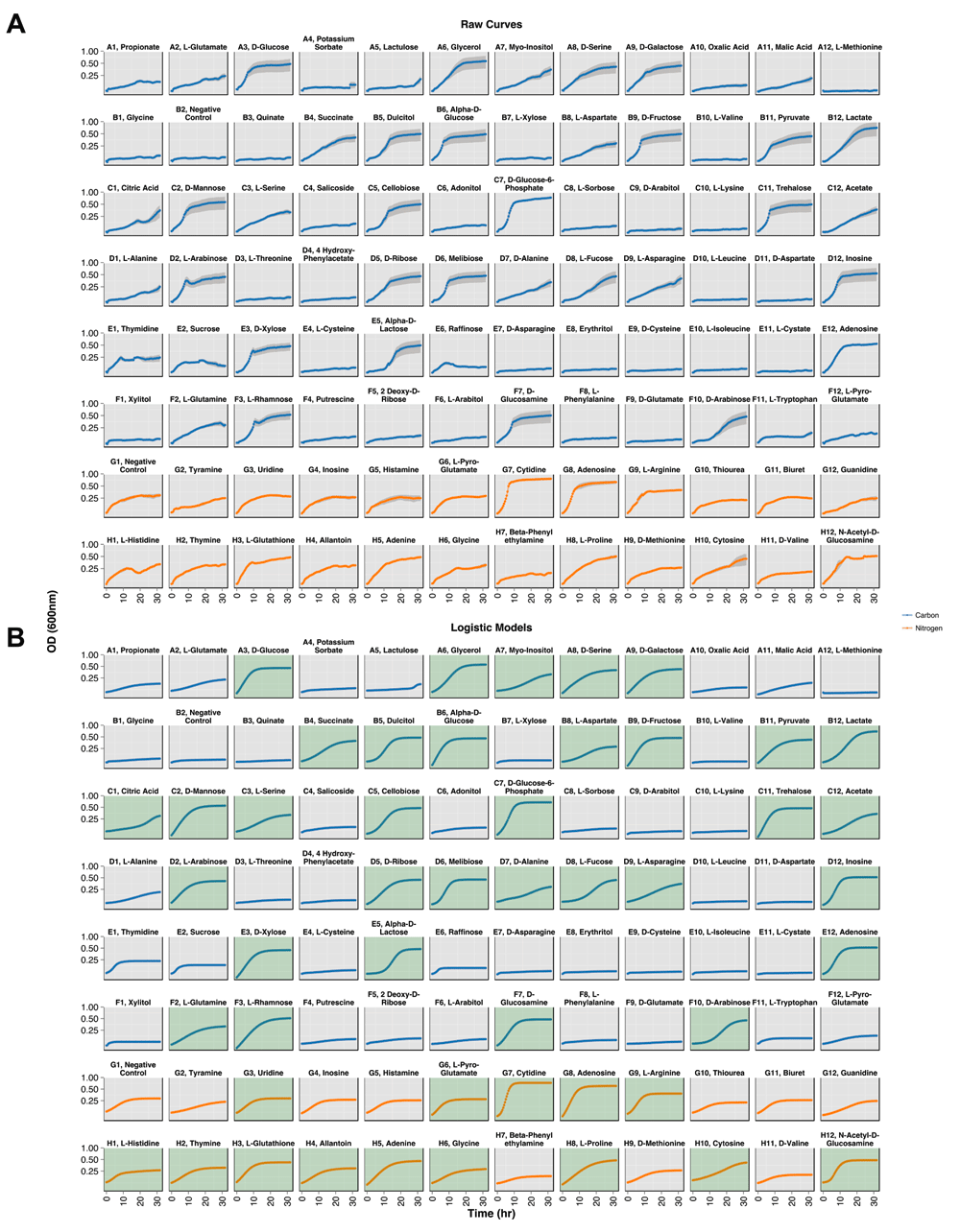
Growth curves generated from the multi-plate reader over 32 hr. The y-axis is displayed in a log2 scale and substrate groups are distinguished by color: blue for carbon sources and red for nitrogen sources. (A) Standard error from the technical replicates are plotted as gray regions. (B) Logistic models of each growth condition. Green panels indicate conditions displaying growth.
Citrobacter sedlakii growth (G) and no growth (NG) phenotypes. Carbon substrates are denoted with (C); nitrogen substrates are denoted with (N). FBA results before gap-filling are shown in the Initial Model column. MAP-FBA comparison results are: correct positive (CP), correct negative (CN), false positive (FP), and false negative (FN).
The initial metabolic model generated in KBase (C.sedlakii_nogapfill) from RAST annotations contained 1,367 reactions and 1,277 substrates. An FBA simulation using this model and specifying LB as the media source resulted in no growth, however only eight reactions were added to simulate growth. These eight were back referenced to the RAST annotations to check for mis-annotations or functional annotations not included in the model. Two gap-filled reactions (dimethylallyl-diphosphate:isopentenyl-diphosphate [EC 2.5.1.1] and geranyl-diphosphate synthase [EC 2.5.1.1]) were mis-annotated by RAST as they both shared a function with another annotated reaction (geranyltranstransferase (farnesyldiphosphate synthase) [EC 2.5.1.10]). Undecaprenyl pyrophosphate synthetase [EC 2.5.1.31] and quinolinate synthase [EC 2.5.1.72] were also annotated and shared a function with the gap-filled reactions undecaprenyl diphosphate synthase [EC 2.5.1.31] and quinolinate synthetase [EC 2.5.1.72], respectively. The fifth reaction (1-deoxy-D-xylulose-5-phosphate pyruvate-lyase (carboxylating) [EC 2.2.1.7]) was not annotated but a comparison between the C. koseri and E. coli K12 genomes revealed neighboring homologs in C. sedlakii genome flanking a gap where the gene should be. Therefore, it is likely that RAST missed this gene during gene calling due to a frameshift sequencing error. Homologs of the proteins for three reactions (ATP:dTMP phosphotransferase [EC 2.7.4.9]; glutamine amidotransferase [EC 2.4.2.-]; riboflavin transport in/out via proton symport) were not found in C. sedlakii but were identified in the C. koseri genome. In addition to the previous eight reactions, three additional reactions (meso-2,6-diaminoheptanedioate carboxy-lyase [EC 4.1.1.20]; NADH:guanosine-5'-phosphate oxidoreductase (deaminating) [EC 1.7.1.7]; prephenate:NADP+ oxidoreductase (decarboxylating) [EC 1.3.1.13]) were altered from being uni-directional to bi-directional. The base model (C.sedlakii_ArgonneLB_gapfill), with the ability to grow on rich media, contained 1,279 (+2) substrates and 1,375 (+8) reactions.
Using the base model, the 90 well simulation resulted in no growth for all 90 growth conditions (53.3% accuracy: 48 false negatives (FN)) (Table 1). Subsequently, gap-filling was performed on those 48 conditions resulting in the reconciled model (C.sedlakii_MOPS_simpheno). The total number of substrates increased to 1,301 (+22 from the base model; +24 total), the total number of reactions increased to 1,399 (+24 from the base model; +32 total), and nine reactions were modified to be bi-directional. Transport reactions made up 11 of the 24 (46%) minimal media gap-filled reactions. When performing gap-filling on multiple conditions, KBase produced a separate solution for each condition. This results in reactions appearing in multiple solutions. To find the missing set of essential reactions, the gap-fill results were parsed to locate the minimum set of reactions present in the majority of the solutions. All new and modified reactions added to the model are listed in Table 2. Although Table 2 presents the gap-filled reactions for only 13 FN conditions, the addition of those gap-filled reactions correctly amended the other 35 FN conditions not shown in the table.
Each gap-filled reaction is listed under their respective media condition, along with the primary source of the compound in parenthesis, i.e. (C) denotes carbon and (N) denotes nitrogen. E.C. numbers were supplied by KBase when viewing gap-filling results. Reactions listed with an asterisk denote the 13 transport reactions added to the model. Reactions listed with a yes in the Reversible column denote those that were already present in the model but made bi-directional through gap-filling.
Mis-annotation checks and cross-referencing with the C. koseri genome were performed for the 12 gap-filled, non-transport, newly added reactions. In four cases (phosphoribosyl-ATP pyrophosphohydrolase [EC 3.6.1.31]; 1-(5-phospho-D-ribosyl)-AMP 1,6-hydrolase [EC 3.5.4.19]; D-mannose-6-phosphate ketol-isomerase [EC 5.3.1.8]; D-lactate dehydrogenase [EC 1.1.2.5]) the DNA sequences were missing but their surrounding genes were sequenced and annotated, indicative of poorly sequenced genes or genes located between contigs. Two reactions (chorismate pyruvatemutase [EC 5.4.99.5]; pyrimidine phosphatase [EC 3.1.3.-]) were mis-annotated by RAST. Four reactions (ureidoglycolate amidohydrolase (decarboxylating) [EC 3.5.3.19]; allantoate amidinohydrolase [EC 3.5.3.4]; allantoin amidohydrolase [EC 3.5.2.5]; 5-oxoproline amidohydrolase (ATP-hydrolysing) [EC 3.5.2.9]) could not be identified in C. koseri and two reactions (D-Arabinose ketol-isomerase [EC 5.3.1.3]; myo-inositol:oxygen oxidoreductase [EC 1.13.99.1]) could not be identified in C. koseri nor in E. coli. Therefore, these six reactions were added with minimal evidence. From the six reactions, three reactions (including the transporter) were required for growth on allantoin. In fact, Citrobacter species have not been shown to be capable of utilizing allantoin as a sole source of nitrogen28. More in-depth experiments are needed to investigate C. sedlakii’s ability to grow in the allantoin-based growth condition.
The 90 simulations were executed again and resulted in growth on 54 conditions (48 TP, 6 FP) and no growth on 36 (36 TN, 0 FN), a final accuracy of 93.3%. The six false positive growth conditions were: 2 deoxy-D-ribose (carbon), glycine (carbon), L-pyro-glutamic acid (carbon), L-threonine (carbon), malate (carbon), and putrescine (carbon) (Table 1). In an attempt to resolve the false positives, the KBase gap-generation algorithm was executed; however, it was unable to identify a set of reactions that could be safely altered or removed from the model without changing correct positive results into false negatives.
Back referencing the gap-filled reactions to the prior RAST annotations provides insight into KBase’s ability to build the initial reconstruction from subsystem annotations. Several reactions identified by RAST were not included in the metabolic model. This is either due to RAST not being able to determine the function of the gene using homology, or the model is not able to correctly incorporate the function from the annotation. Gap-fill on LB media identified four reactions that were later categorized as RAST mis-annotations, whereas gap-fill on the MAPs media identified only two reactions later categorized as RAST mis-annotations. By surveying neighboring homologs of gap-filled reactions in closely related genomes, one reaction on LB gap-fill and four reactions on MAPs gap-fill were categorized as missing due to poor sequence quality.
As more bacterial genomes are processed through this pipeline, mis-annotations will occur at a lower rate. In the case of C. sedlakii, six reactions out of the 19 non-transport, newly added reactions were missed due to mis-annotations; five reactions possibly located in-between contigs; eight not bearing homology with related organisms. Feedback from mis-annotations corrects the ambiguities for future bacteria metabolic reconstructions as administrators of KBase, RAST, and the SEED database are informed of these findings. Reactions that are gap-filled and not identified as mis-annotations will be recorded for future studies. Patterns of missing reactions provide insight into why RAST is not able to identify the functions from an organism’s sequences. Furthermore, closer investigation can answer if the gene encoding the protein for a particular species (or genus) does not share strong homology to its closest evolutionary neighbor.
Thirteen out of the 32 essential reactions (41%) added during gap-filling were transporters. For specific conditions, the only missing reaction that prevented growth during the simulation was observed to be transport proteins specific for the growth condition (see Table 2). Transporters are readily identified using homology-based searches, however, it is difficult to accurately identify which substrate(s) are actively transported using these techniques. A drawback of RAST is its dependence on sequence homology. Henry et al.30 indicated that poorly annotated transporters are typically missing from preliminary reconstructions using the SEED database. During metabolic reconstruction in RAST, a minimal set of reactions are uniquely chosen through an optimization equation. The equation contains a penalty parameter that favors intracellular reactions over transporters during the auto-completion step of the model reconstruction, thus further preventing transporters from being included in the draft model.
False positive (FP) results were introduced during the reconciliation process. A false positive was defined as an FBA resulting in biomass production in nutrient conditions where C. sedlakii is not able to produce biomass, i.e. the MAPs assert no growth. FPs are an indication of an under-constrained model that has resulted from the addition of reactions or from making reactions bi-drectional, both of which are enabling biomass production during FBA. In the case with the C. sedlakii model, FPs resulted after the gap-fill occurred. The 32 reactions added and nine reactions re-labeled as bidirectional have enabled growth in six growth conditions where the MAPs display no growth. KBase function fba-gapgen attempts to correct these issues. To perform this function, parameters pertaining to the growth condition where the FP occurs and a growth condition where a correct positive occurs are required, allowing the algorithm to correct, or remove, reactions from the model without altering the outcome of the correct positive growth condition. The KBase software was unable to determine any reactions in our model to remove with the fba-gapgen function, suggesting that the model requires manual curation and in-depth experimentation in order to resolve these six FP growth conditions listed in Table 1. Certain biochemical events not captured by the metabolic model may be occurring, such as the effect of transcription repression and other complex cellular behaviors. FBA alone is unable to account for such dynamic responses, although efforts have been made to extend genome-scale metabolic models with metabolic and gene expression data34,35.
Models created using the discussed pipeline should be considered draft metabolic models. While the physiological experiments provide insight into an organism’s metabolic capabilities for substrate utilization and the subsequent biomass formation, the biochemical properties involved are not directly assessed. The gap-filling algorithm attempts to determine the minimal set of reactions required for growth for a specific condition but these are not considered finite decisions. Gene knockout experiments, extensive literature mining, and manual curation are required to enhance the models31. However, these alternatives are neither high-throughput nor considered in this pipeline but are encouraged for further studies. Cross-referencing to closely related organisms can give insight into the validity of adding a reaction to the model28.
The prevalence of complete and near-complete draft genomes is increasing as DNA sequencing becomes cheaper and more robust. Unique bacterial species are now being studied and their data is becoming more readily available for interpretation. We describe a process to combine DNA sequence data and phenotypic experiments to produce a programmatic metabolic model construct. Metabolic reconstructions are built using RAST genomic annotations by implementing PMAnalyzer, a high-throughput pipeline. Biochemical reactions not captured by homology-based algorithms are highlighted using flux-balance analysis and gap identification techniques hosted openly on the KBase platform. Citrobacter sedlakii was used as a model organism to describe, test, and critique the pipeline. FBA results using this model were shown to achieve a 93% prediction accuracy, an improvement from the initial model providing a 53% prediction accuracy.
The high-throughput pipeline presented combines physiological data with genomic information to result in more accurate metabolic models. Using experimentation to validate model prediction and improve model capabilities has been shown previously, however, these processes are not streamlined to be fast or robust. Our methodology implements speed and robustness at every level of the work-flow. Using a multi-plate spectrophotometer, hundreds of different growth conditions targeting several metabolic pathways results in an expansive phenotypic profile. The PMAnalyzer pipeline executes rapidly and is integrated into an automated web server where users obtain phenotypic results within minutes. Draft model construction and model reconciliation on KBase are completed quickly, providing gap-filled biochemical functions of a genome to use in subsequent functional analyses.
figshare: Phenotypic profiling data for elucidating genomic gaps. Doi: 10.6084/m9.figshare.396907232
Latest Software script source code: https://github.com/dacuevas/PMAnalyzer
Source code as at the time of publication: https://github.com/F1000Research/PMAnalyzer/releases/tag/V1.0
Archived source code as at the time of publication: http://dx.doi.org/10.5281/zenodo.1141333
Software License: GNU GPL v3.0
DAC and RAE conceived and designed the study. DAC wrote the paper, prepared figures and tables, developed and enhanced the PMAnalyzer code and pipeline, designed the experiments, performed experiments, and analyzed the data. DAC, DG, SES, and JR jointly discussed the design of the PMAnalyzer. SES designed the MAPs and performed the MAP experiments. ED provided DNA sequencer instrument, reagents, and sequences. JR, AS, and FR included helpful discussions with the MAPs. CSH contributed code to KBase. VV and RAO provided helpful discussions and suggestions with genome annotations.
This work is partially supported by NSF grants CNS-1305112 and MCB-1330800 to Edwards, DUE-132809 to Dinsdale, DEB-1046413 to Rohwer, and by a STEM scholarship award funded by NSF grant DUE-1259951 to Cuevas.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
A special thank you to Barbara Bailey, Ben Felts, Jim Nulton, and Peter Salamon from the San Diego State University Bio Math group for their discussions and opinions with bacterial growth curve modeling.
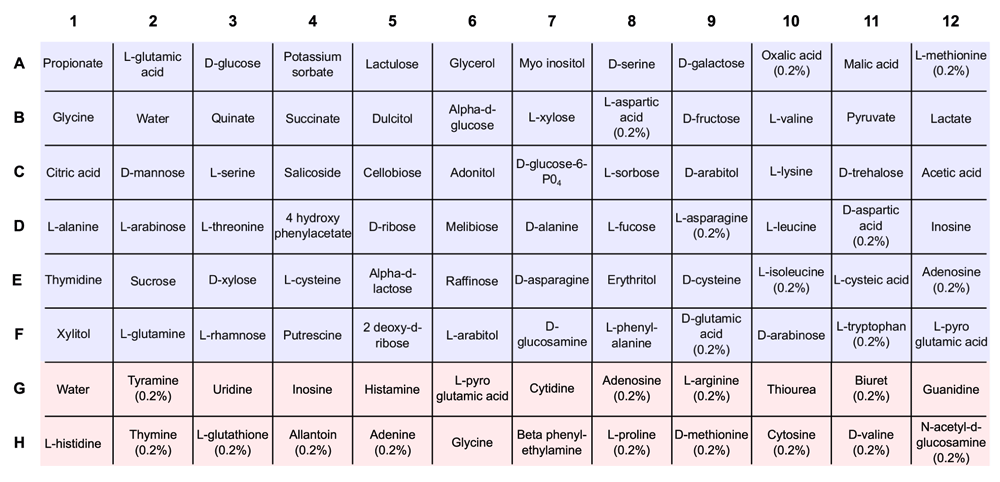
Each well contains 60 µL of sterile water, 50 µL of 3X MOPS basal media, and 30 µL of 5X substrate. Unless noted, a concentration of 0.25% is used for each substrate. Carbon substrates are denoted by blue squares in rows A–F; nitrogen substrates are denoted by red squares in rows G and H. Water denotes wells using water instead of a carbon (or nitrogen) substrate.
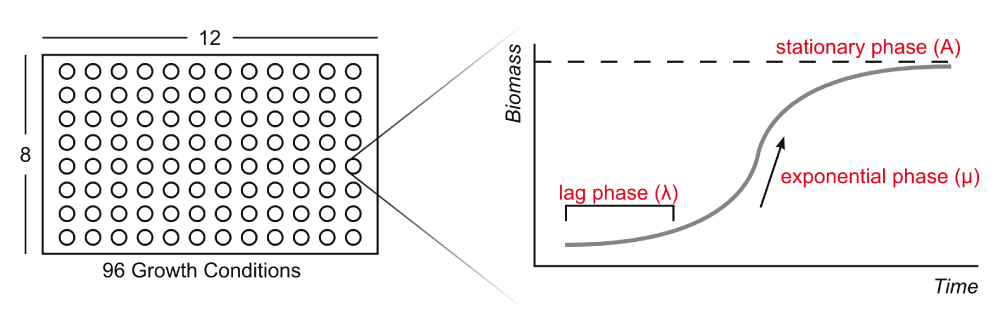
A bacterial growth curve can be parameterized into three phases: lag phase, exponential phase, and stationary phase. Parameters correspond to the logistic equation described in the text (Equation 1).
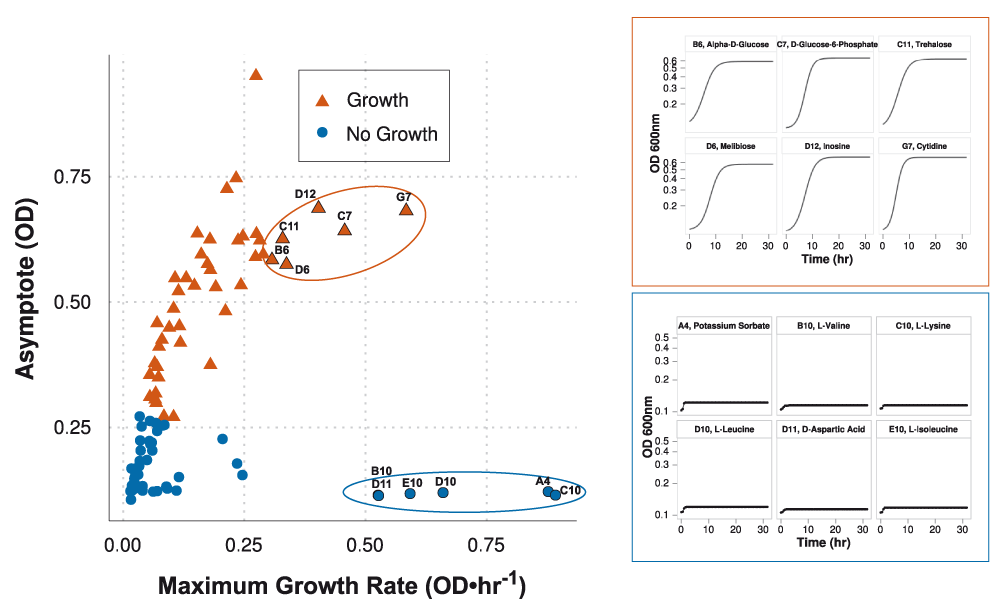
Classification of growth conditions are based on the asymptote adjusted logistic model calculation in Equation (4). Red triangles and blue circles indicate conditions asserting growth (≥ 0.5) and no growth (< 0.5), respectively. Classification is weighted by the final biomass yield (asymptote) rather than growth rate. Maximum growth rate can be misleading; e.g., growth curves highlighted in the blue box were modeled using high growth rates but do not assert growth. Growth curves highlighted in the red box were modeled using slightly lower growth rates but assert growth.
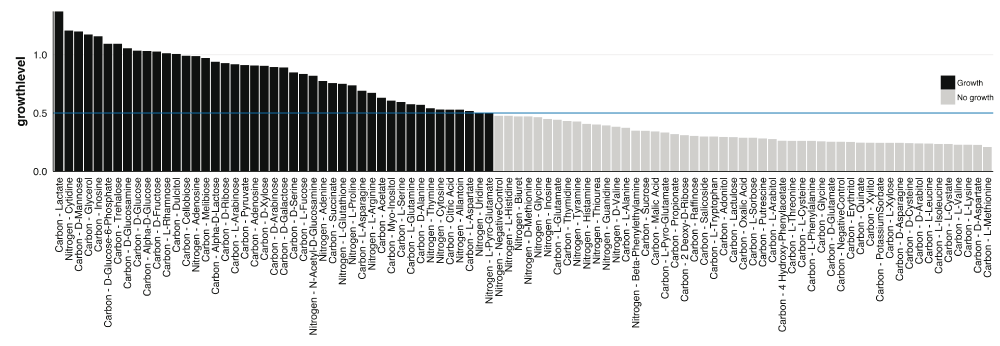
Growth level values for each condition. Blue horizontal line represents the threshold used for defining growth and no growth.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)