Keywords
Sequence alignment algorithms, hydrophobicity scale, protein homologs, TMATCH
Sequence alignment algorithms, hydrophobicity scale, protein homologs, TMATCH
An understanding of the properties and functions of a protein or a nucleic acid often begins with a search of the sequence against databases of proteins (or nucleic acids) with known properties or functions. The fundamental assumption is that sequence leads to structure which in turn leads to an understanding of the function. Search algorithms have improved and continue to improve. Yet, with proteins in particular, it remains difficult to detect remote homologies in the so called twilight zone where proteins have low percent sequence identities starting around 20–25% and descending to around 10–15%. We describe a hydrophobicity scale that is proving to be an excellent measure of sequence relatedness. A robust estimate of the hydrophobicity based sequence identity can be calculated directly from a global alignment score, which may be directly used in database searches. Proteins with low sequence identities, possessing statistically insignificant similarities by conventional measures, but having similar secondary/tertiary structures, which would not be identified as statistically significant by other methods such as FASTA and Smith-Waterman can be identified as homologous using our new alignment algorithm (unpublished report) through the enhanced information content of our hydrophobicity proclivity scale.
Hydrophobicity scales (metrics) as understood in the literature are generally divided into four categories, derived from
Experimental physio-chemical data
Log of a partition coefficient derived from protein structure (e.g. Fraction amino-acids inside vs. outside, fraction amino-acids in contact with water vs. completely buried, etc.)
Amino-acid mutation/substitution rates and
Participation rates/probabilities of occurrence in folded protein secondary structure
There are a large number and myriad types of scales that appear in the literature starting from the 1960’s through to the present with a fair amount of variation amongst these scales. The correlation between some of the hydrophobicity scales can be best understood as that derived from the energy of interaction between amino-acids and water or the energetics of partition of amino-acids from water as the reference state and some other environment such as a non-polar solvent or the interior of a folded protein. Hydrophobicity can thus be joined within a single, unified, conceptual framework1,2 Through extensive analysis (primarily using regression and scatter plots), we were able to identify patterns and arrive at metrics describing amino acid properties. We derived a number of additional metrics by differentiating metrics that were intrinsic as opposed to extrinsic, as understood in thermodynamics. Extensive cross correlation with the primary and derived metrics using regression modelling were undertaken to recover the best and most meaningful hydrophobicity metrics. We relied on several different sources for our analysis. For data on amino acid surface areas, we used Rose et al.3. Amino acid mass information was obtained using the AAINDEX accession number #FASG7601014,5. Amino acid volume data was obtained from Creighton6 Amino acid absolute entropy of formation was from the AAINDEX database using accession number #HUTJ7001024,5.
We arrived at our hydrophobicity scale after exhaustive analysis which included numerous scatter plots and the running of a number of multiple regressions. The question we were trying to answer was - What was the best hydrophobicity scale, or combination of scales, that best represented the role of the different amino acids in proteins?
We started by first collecting many hydrophobicity indices and physico-chemical indices from the literature and scatter plotted/regressed the hydrophobicity indices against each other, and the harvested physico-chemical properties and their derived intrinsic properties of amino acids. For example when a hydrophobic scale is plotted against the ratio of the surface area per specific volume (volume/molecular weight) of each amino acid we get a scatter plot with a distinct pattern. In such a scatter plot, we can identify one or more sets of linear clusters of amino acids, each set of which is considered to be a “property class”.
Consider Figure 1 where our normalized average hydrophobicity index is scatter plotted against the area per specific volume of each amino acid (shown using their alphabetical representations).
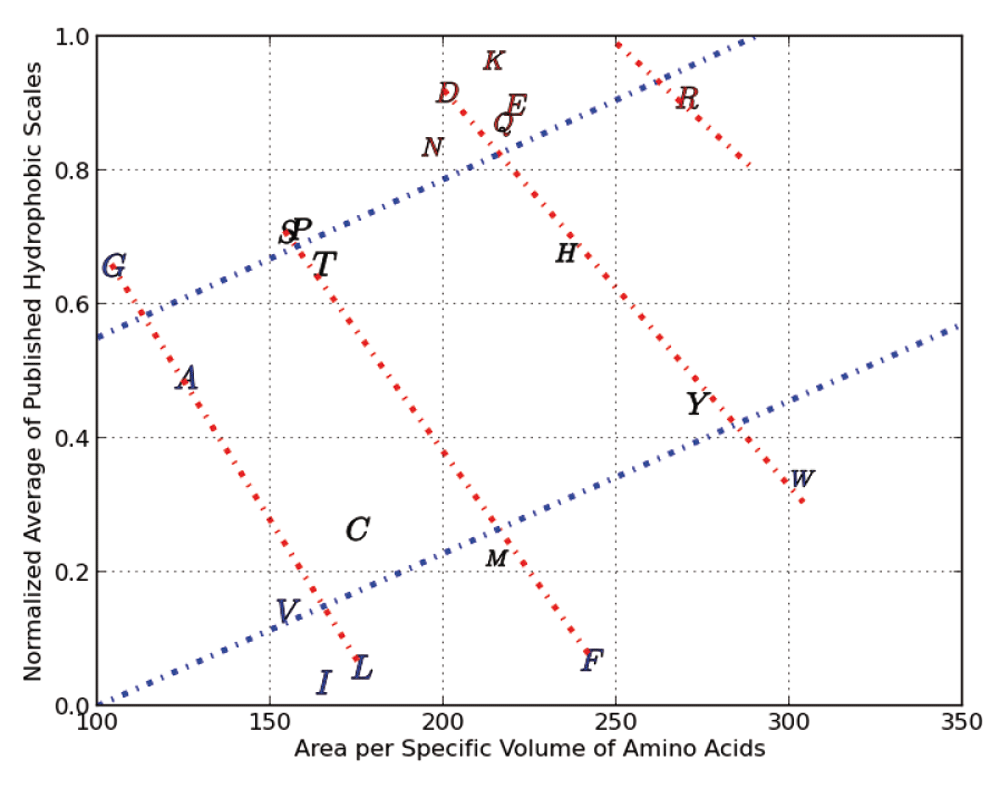
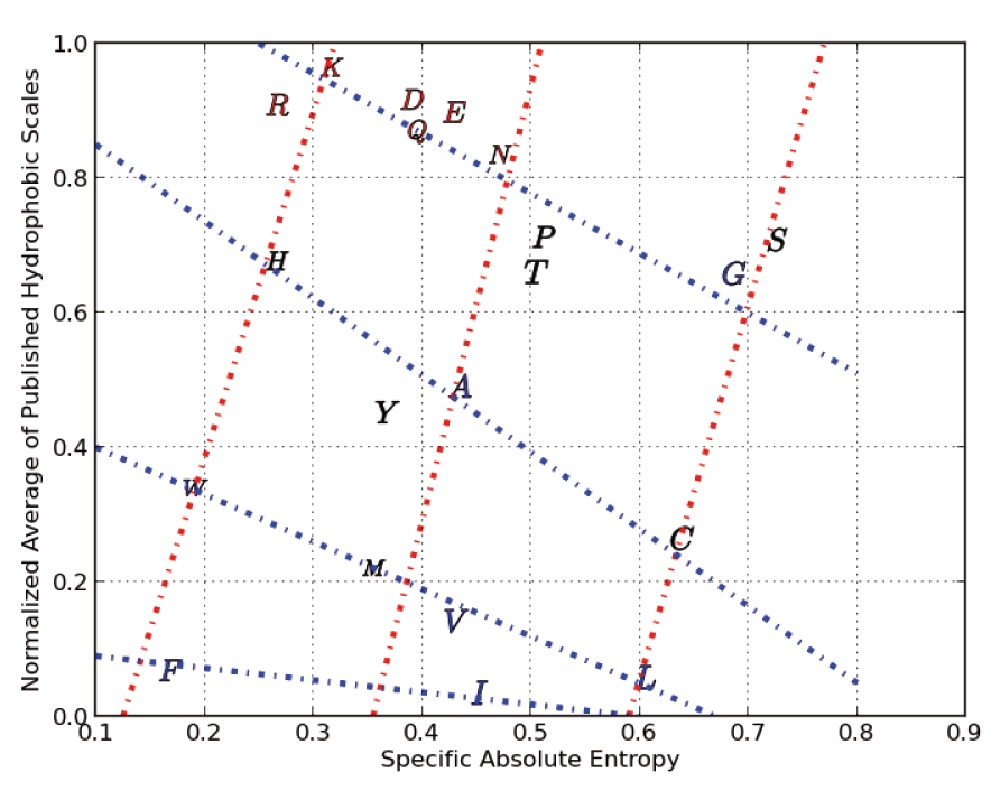
We can clearly see cross-hatched patterns where for example the amino acids G, A, C, V, I and L are on a straight line (starting from the top left to bottom right). Moving right, we see that S, P, T, M and F are on a straight line (nearly parallel to the line formed by G,A,C,V and I). Continuing further right, we see a third line which crosses several amino acid, followed by an outlier, amino acid R. This series of four lines form what we call Property Class 1. We assign a numerical value of 0 to the line through G,A,C,V and I and a value of 1 to the next line and so on. In the same Figure 1 we can see the formation of Property Class 2 which contains only two series. We arrived at Property Class 3 and Property Class 4 by scatter plotting our normalized average hydrophobicity index against specific absolute entropy (and this is shown in Figure 2) The four property classes we identified respectively in the scatter plots shown as Figure 1 and Figure 2, along with the respective X axes physico-chemical property, correlated very highly (as multiple linear regression factors) with our normalized average of three robust hydrophobic indices’s (shown as ave3H) having an R squared >95%.
Property class #5 reflects a scatter plot between the delta G of burial of AA secondary groups7 (as Y) and the number of atoms in the respective secondary group6, which resulted in 5 linear series. Each of the linear series numbers (0 through 4) for each AA forms the basis of property class #5. The multiple linear regression of the delta G of secondary group burial with number of secondary group atoms and property class #5 resulted in an R2 of 98.1%. Property classes #6, #7 and #8 were derived from 49 fundamental amino-acid properties and derived scales that are based upon an analysis with Analysis of Patterns (ANOPA)8. Together PC #1 to #8 represents 8 X vectors (listed in Table 3) in the multiple linear regression reported in the third column of Table 2. The property class index vectors are shown in Table 3.
We were able to find three hydrophobicity scales that were the most robust from the regression cross correlation study. The hydrophobicity proclivitity scale that we report in the present paper are the normalized average of three normalized scales2,9,10.
Our hydrophobic index is the result of an extensive mining of the literature about proteins and amino acid scales/metrics in different environments. Almost all hydrophobicity scales reflect in some way a measure of the energetics of transfer of an amino-acid (or proteins) from one solvent environment (water) to another (folded protein or multiple protein assembly). During our data mining and analysis, three hydrophobicity metrics emerged as the most appropriate since we could relate those scales to multiple fundamental properties of the 20 natural amino acids using multi-variate statistical procedures, thermodynamics and biophysical chemistry considerations2,9,10. Hydrophobicity scales reflect different physical properties of amino-acids, such as metrics derived from amino-acid partitioning patterns (e.g. from the hydrophobic core to the exterior of proteins) or log of partition ratios between water and organic solvents. We found, as widely suggested in the literature, that the free energy of transfer from water to octanol turns out to be a good proxy for the hydrophobic core environment of folded proteins.
We created a normalized average of the three key hydrophobicity scales (The index i=1 is from Tang2, index i=2 is from Neumaier9 and the index i=3 is from the average of the collected scales in Juretic10). This normalized average of three scales provides a reasonably unbiased estimate of the "true" average hydrophobicity relationship amongst the 20 amino-acids (index j, from 1 to 20)
The hydrophobicity scale as calculated using Equation 2 using the scales published in2,9,10 has a number of interesting relationships with key physico-chemical properties of the amino-acids in proteins. For example, this normalized average of these three best hydrophobicity metrics possesses statistically significant linear correlation with many other reliable hydrophobicity metrics derived from multiple literature hydrophobicity scales.
An example scale, derived from an analysis of 28 literature hydrophobicity metrics, possesses a strong linear relationship (R2 = 0.959) with our normalized average of three hydrophobicity scales, that forms a hydrophobicity proclivity scale, has been published in 1.
Hydrophobicity scales are typically derived from a measure of the probability that a particular residue will be buried in the core of the protein, away from water. What confounds these calculations is the fact that in most proteins, many of the hydrophobic residues are still exposed to the water (solvent). It is often not clear on how to treat residues that have properties intermediate between hard core hydrophobic and polar residues. The size of the residues and difference between alkyl and aromatic residues also pose some difficulty in the calculation of a hydrophobicity scale. Calculations involving cysteine residues add additional complexity in that some of those residues may be involved in providing proteins structural stability through formation of disulfide bonds. Thus, calculation of contributions to any hydrophobicity index through analysis of where specific residues are in a given protein has been complicated and contributed to the scatter we see in the data. We demonstrate this by examining the normalized average of several popular hydrophobicity scales11–16 versus the probability of an amino-acid solvent-exposed area (SEA)17,18 greater that 30 (shown in Figure 3)
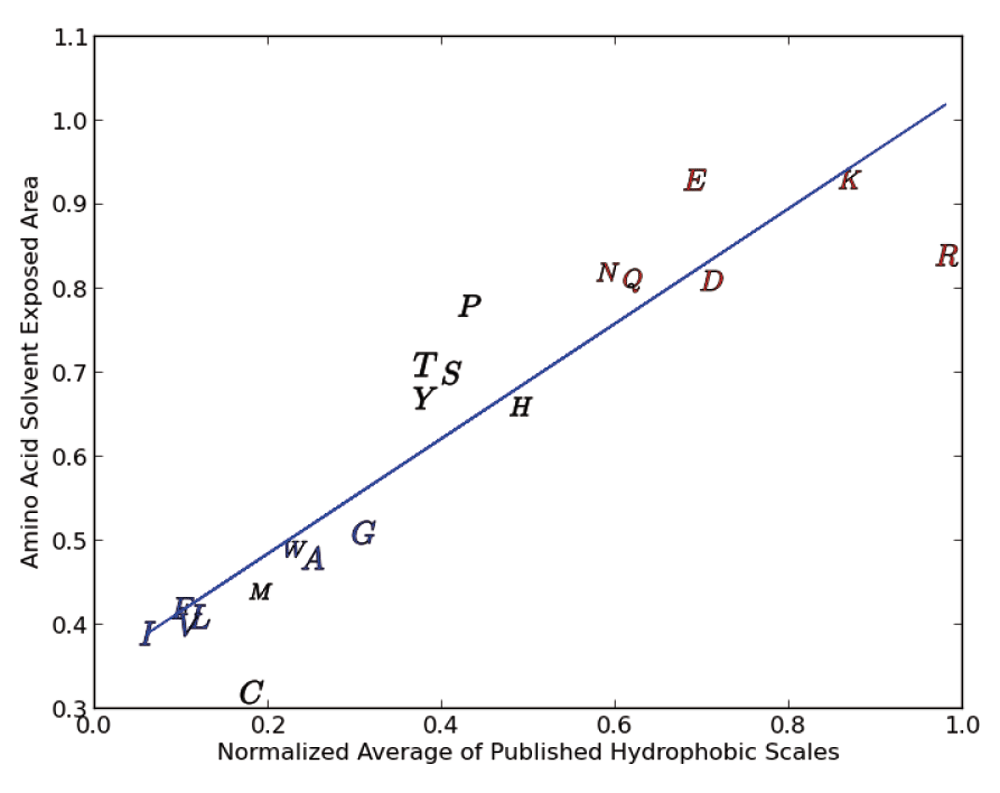
Figure 3 shows that there is indeed a relationship between the hydrophobicity scale and whether or not a particular amino acid is within a protein core or exposed on the surface. We see one tight grouping of amino acids in the figure (I, F, V, L, M, W, A and G) and two loose groupings that include P, T, S, Y, H and N, Q, E, D, K and R. The group at the top right (N, Q, E, D, K and R) include amino acids that are ionic/strongly polar and the central group of amino acids are of intermediate polarity. The tight group of amino acids are primarily amino acids with hydrophobic residues. As we go from the very hydrophobic group to the less hydrophobic group (from the lower left to the top right) the scatter goes up. This scatter is indicative of the increase in water amino acid interaction and of the difficulty of accurately calculating the contribution of any particular residue.
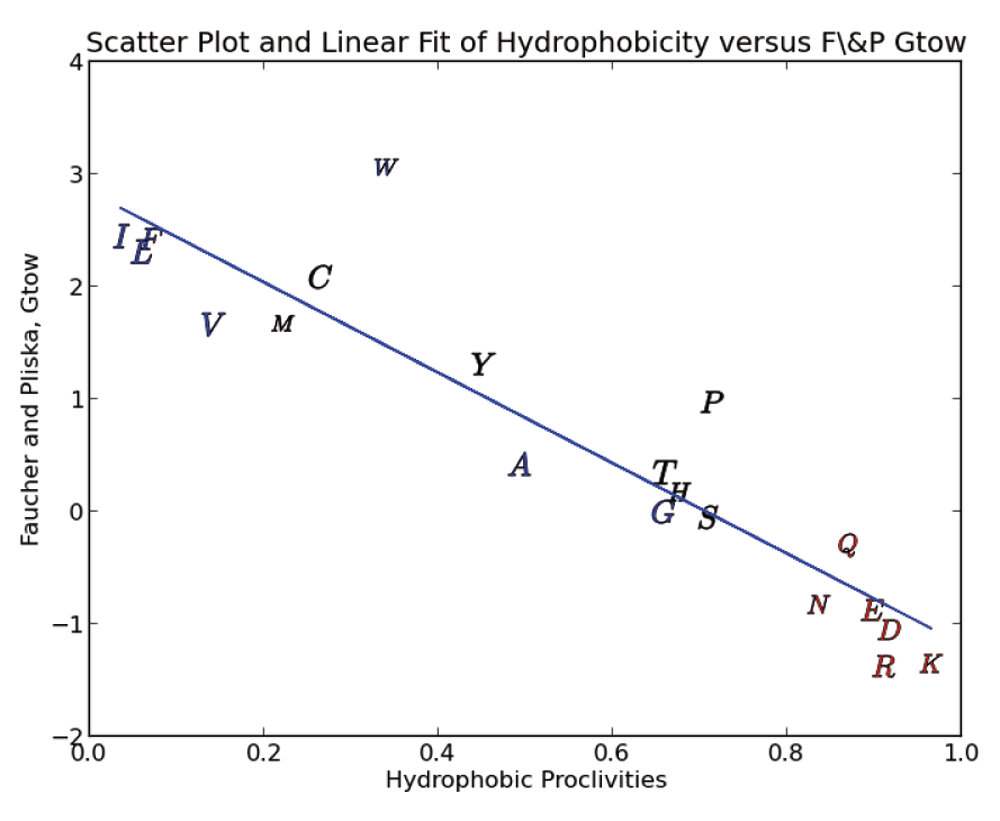
In Figure 4 we show a scatter plot of our amino-acid hydrophobicity proclivities against the popular Fauchere & Pliska free energy of amino-acid transfer from n-Octanol to water (Gtow) scale7,19. It is common in the literature to see n-Octanol used as a proxy for the typical hydrophobic core of folded globular proteins, consequently the Gtow scale has been widely used as a measure of hydrophobicity. As can be seen above the correlation is quite good at 85.9% linearity (coefficient of determination). The regression of these two scales is used to derive a fitted free energy of transfer and reported in Table 1 and used in our new alignment algorithm. Since Gtow reflects a delta G (energy) of transfer, hydrophobic proclivities can also be seen to relate directly to energy.
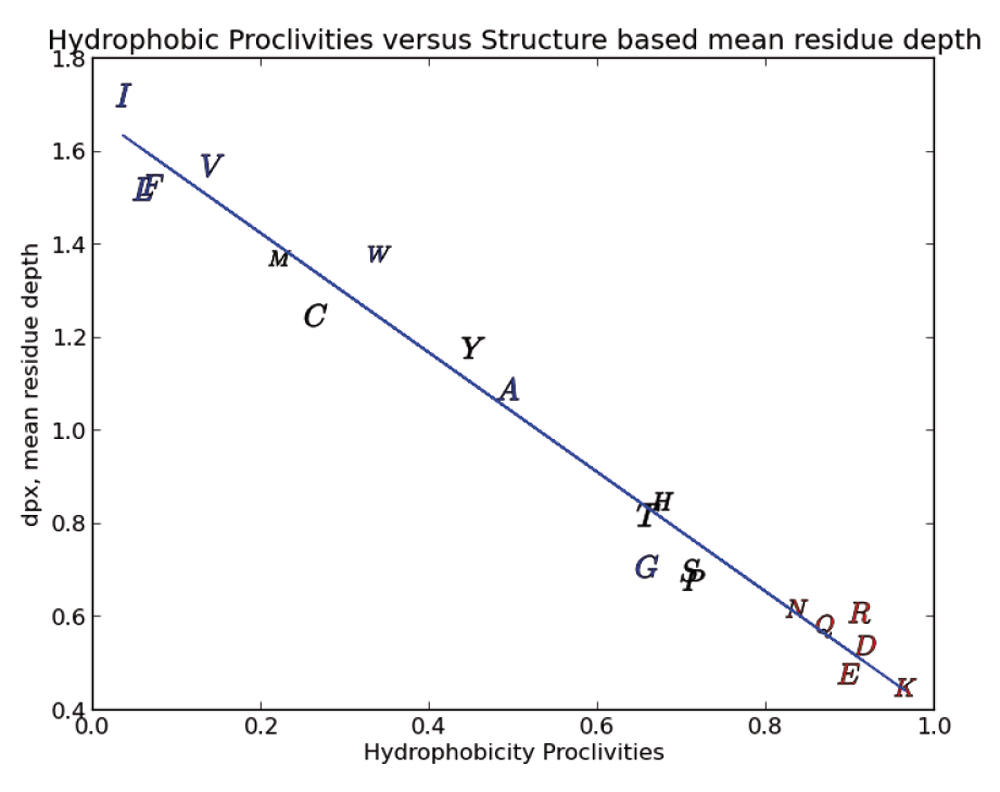
The reasonableness of our hydrophobicity scale is also demonstrated by examining the relationship between our scale and the mean residue depth (dpx) defined as the distance between the interior of a protein amino-acid and the nearest water molecule in the aqueous shell surrounding the protein20,21. In Figure 5 we show that there is a strong relationship (97% linearity) between the dpx metric and our hydrophobic proclivities. The dpx metric is a straight forward geometrical description of the local protein interior and can be expected to provide similar information to the solvent accessible area and buried surface area metrics. The dpx depth and hydrophobic proclivities correlate with amino-acid/protein properties such as average protein domain size, secondary structure, protein stability, free energy of formation of protein complexes, major literature amino-acid hydrophobicity scales, residue conservation, post-translational modifications like phosphorylation, and hydrogen/deuterium amide proton exchange rates7,20,21.
In Table 2 we summarize the performance of several of the hydrophobicity scales published in the literature. The hydrophobicity scales shown as rows are compared with four important quality metrics that are either amino acid physico-chemical properties or derived from such properties. The quality of inter scale regressions are shown as R2. The performance of each row scale can be observed relative to the other row scales within each of the four columns, where the higher the R2 the better the performance of the row scale with regards to the column scale. There are 13 rows in Table 2 representing 11 hydrophobicity scales, one solvent exposed area scale and one delta G of transfer from water to an organic solvent (Octanol).
Of the 11 hydrophobicity scales in Table 2, 7 are popular scales in practice, three are the constituent scales of our hydrophobicity proclivity scale and our hydrophobicity proclivity scale. These row choices in Table 2 are to illustrate a close relationship between AA hydrophobicity and the transfer of an amino acid to an organic solvent (n-Octanol, column 2), used as a proxy for the internal environment of a folded protein, as well as to compare AA hydrophobicity with an AA Solvent Exposed Area scale (column 1) also representing a folded protein environment. The high R2 between the row dG of transfer to Octanol and the first column AA Solvent Exposed Area (SEA) scale in Table 2 illustrates the aptness of comparing the dG of AA burial in protein "solvent" to a solvent-solvent transfer model between water as the reference state and an organic solvent as the transfered or final state. In Table 2, the inclusion of the row SEA is to illustrate the high R2 with the first column SEA illustrating the consistency of folded protein behaviour in SEA scales derived from different data sets. With the Rose AA percent buried row hydrophobicity scale3, simlar lessons can be gleaned as with the row Octanol and SEA scales, as the Rose scale represents the environment of a folded protein. The very high R2 between these three row scales and the last two column regression scales in Table 2 illustrate a strong justification for including these row scales, as protein folding is thereby strongly linked with other physico-chemical properties of amino-acids, as reflected by these two columns. We describe the regression X variables in the 4 columns of Table 2) below.
We can see that the correlation between our hydrophobicity scale (shown as avg 3H in Table 2) and the Moelbert average amino-acid solvent Accessible Surface Area (ASA) within proteins has an R2 = 84.7%. The ASA is the average area of each amino acid exposed to water in the globular proteins. When our hydrophobicity proclivity scale approaches 1 (i.e. hydrophilic) the ASA goes up as would be expected, with the converse being true as our hydrophobicity scale approaches 0 (i.e. hydrophobic) the ASA goes down22.
The amino-acid Accessible Surface Area (ASA) has long been suggested as a reasonably accurate proxy for hydrophobicity7,17,22 as is also seen in a related scale, the Solvent Exposed Area > 30 square angstroms17,18. The amino-acid property classes are vector sets of clusters/linear families of curves in multiple linear regression relationships between two (or more) amino-acid physico-chemical properties. The first two columns (ASA and Gtow) represent paired variable linear regressions and the third column (Property Classes #1 to #8) and fourth column (Property Class #1 to #4, AA area/specific volume6 and specific absolute entropy4,5) represent multiple linear regressions.
The R2 in the first two columns of Table 2 represent linear regression results between the Y (row) vectors and the X (column) vectors. The R2 in the last two columns of Table 2 derive from multiple linear regressions, where the independent (X) variables are vectors of amino-acid property Classes (PC) and/or amino-acid physico-chemical properties, and each row parameter is the dependent variable, respectively. Again, the Property Classes can be thought of as distinct subsets of amino-acids representing multiple linear series/clusters (within scatter plots or multiple linear regressions) of amino-acids in reference regressions associated with X variable vectors from some key physico-chemical metrics plotted against the hydrophobicity proclivity vector scale.
In Table 2, we see that the F and P Gtow scale performs as well (i.e. high R2) as the best of the hydrophobicity scales within columns 1, 2 and 4, thus, further justifying our selection of the Gtow scale as our baseline standard for a free energy of transfer from an aqueous solvent environment to a non-aqueous solvent. The SEA > 30 A2 does as well as the popular hydrophobicity scales in Table 2 and has good correlation with the F and P Gtow scale in column two and thereby establishes a direct link between the F and P Gtow scale and the free energy of burial of amino-acids in proteins and providing strong evidence justifying a solvent-solvent transfer model for protein folding.
The Tang Q and Neumeier X scales are the top performing individual hydrophobicity scales as seen in the first two column results, followed on average by the Rose scale. The Juretic Avg scale generally performs as well as the five popular hydrophobicity scales in columns one and two, but more importantly it performs better than any other single hydrophobicity scale except for the Tang Q and Neumeirer X scales in columns three and four. Since we consider columns three and four to be a more rigorous test for a robust, high performance hydrophobicity scale, we see the justification for selecting the Tang Q, Neumeirer X and Juretic Avg as the scales from which to prepare our hydrophobicity proclivity (3H) scale. Our hydrophobicity proclivity scale performs basically as well as the best individual hydrophobicity scales in columns one and two, but it is the top performer in columns three and four. No other hydrophobicity scale that we evaluated on average performed as well (i.e. magnitude of R2) in regression comparisons with amino-acid physico-chemical properties as our hydrophobicity proclivity scale.
In Table 2 column three is the 8 sets of numbers (vectors), dubbed as property classes and are eight X vectors in the multiple linear regression relationships with the R2 shown in the third column. These eight property class vectors can form multi-linear regression fits with very high R2 with a large number of the physico-chemical properties of the of the 20 amino-acids in our accumulated AA physico-chemical property database, thereby serving as proxy’s for these properties. In Table 2 column four, we see four property class vectors (#1-#4) and two AA physico-chemical property vector scales (surface area/specific volume, specific absolute entropy); column four is included to illustrate the method of construction of the eight Property Class (PC) vectors represented by column 3.
The great organizing principle embodied within the hydrophobicity proclivities (and implied by dpx), is that of a neo solvent-solvent partitioning effect, where the energetics of the solvent shell waters are the dominant effect in the energy balance. As with clathrates (ordered aqueous shells), which form spontaneously with hydrophobic molecules, there is a solvent shell of ordered waters that form spontaneously around solvated globular proteins. However, there is a confounding factor in trying to obtain an accurate hydrophobicity proclivity in that even the most hydrophobic residue will have some average solvent exposed area, so it is reasonable to postulate that there is some functional reason for exposure of some grease to the solvent The presence of hydrophobic surface area causes an aqueous clathrate shell to form at that point perhaps effectively becoming part of the folded structure of the folded protein, possibly as a retaining structural element operating through surface tension and putting the interior of the globular protein under pressure.
The importance of amino-acid hydrophobicity to the structure and function of globular proteins is critical to the function and survival of cells, a reality that is even reflected in the very structure of the standard genetic code. The amino-acid codons are arranged/coded in such a way as to reflect the underlying hydrophobicity of the respective amino-acids. A careful analysis reveals that the genetic code has a built in redundancy through amino-acid hydrophobicity (in addition to codon redundancy) such that point mutations in a codon that yield a different codon tend to result in an amino-acid with similar hydrophobicity. It has been shown that the underlying amino-acid codon structure has a direct relationship with high quality hydrophobicity scales that are published in the literature23.
A legitimate question about the hydrophobic proclivity scale we have described is why our scale is superior to alignment score matrices such as PAM (Point Accepted Mutation)24, BLOSUM (BLOck SUbstitution Matrix)25 or Gonnet26 that continue to be used for multiple protein alignments and database search alignments. There are indeed several practical and theoretical problems with the use of these log odds score matrices for the alignment of divergent protein sequences. For example, BLAST and several of the major multi-sequence alignment programs like Clustal W use particular BLOSUM matrices as the default. BLAST uses BLOSUM62 as the default. Quotes from select papers have been summarized below to more clearly illustrate these problems.
The substitution matrices used by the alignment programs are generally log of Bayesian probabilities for two amino-acids I and J of the form:
The probability of occurrence of the 20 primary aminoacids is not the same throughout the domain/kingdoms of life, so this mathematical formulation can cause issues for identifying and aligning homologous proteins.
Superimposed on the log of Bayesian probabilities formalism are evolutionary models derived from Markov stochastic process evolutionary models (PAM), which implies apriori knowledge of the evolutionary amino-acid substitution rates. Necessarily, if one chooses PAM or BLOSUM, one must choose one of the series of matrices that one believes is appropriate for the approximate evolutionary distance between any two protein sequences under analysis. Obviously, this practice can cause an undue restriction if the evolutionary distance is too great within the protein dataset being aligned. The only assumption that we make with hydrophobicity and our new alignment algorithm is that nature will strongly tend to substitute similar amino-acids in order to preserve the overall function and structure of homologous proteins, and that it is possible to define a hydrophobicity distance to define a fuzzy match between any two amino-acids, which is recognized as a “similarity match.”
We summarize the salient points regarding alignment matrices with quotes from four select literature articles below.
1. “The most common substitution matrices currently used (BLOSUM and PAM) are based on protein sequences with average amino acid distributions, thus they do not represent a fully accurate substitution model for proteins characterized by a biased amino acid composition”27
2. “We have investigated patterns of amino acid substitution among homologous sequences from the three Domains of life and our results show that no single amino acid matrix is optimal for any of the datasets”28
3. “Many phylogenetic inference methods are based on Markov models of sequence evolution. These are usually expressed in terms of a matrix (Q) of instantaneous rates of change but some models of amino acid replacement, most notably the PAM model of Dayhoff and colleagues, were originally published only in terms of time-dependent probability matrices (P(t)). Previously published methods for deriving Q have used eigen-decomposition of an approximation to P(t). We show that the commonly used value of t is too large to ensure convergence of the estimates of elements of Q. We describe two simpler alternative methods for deriving Q from information such as that published by Dayhoff and colleagues.”29
4. These authors note another interesting problem with the residue substitutions rates use in the Q matrix: “Because different local regions such as binding surfaces and the protein interior core experience different selection pressures due to functional or stability constraints, we use our method to estimate the substitution rates of local regions. Our results show that the substitution rates are very different for residues in the buried core and residues on the solvent-exposed surfaces.”30
Tomii et al.5 essentially conclude that in the “evolutionary” limit, alignment/mutation matricies reflect the hydrophobicity and amino-acid secondary group size. For example, when the correlation coefficient between a hydrophobicity scale and a amino-acid secondary group size, and the PAM matricies are plotted against the PAM distance, the correlation coefficient monotonically increases from 0.58 at a PAM near zero, to a PAM distance of 200 where the correlation coefficient reaches an asymtotic limit of about 0.735.
The amount of information available to an alignment algorithm is essential to its ability to find matching proteins, especially matches with remote homologies where the percentage identity has dropped off to around 20–25%. In this study we have sought to find an optimalhydrophobicity scale that would reflect the real properties of amino-acids within the context of folded proteins. We contend that hydrophobic proclivities transcend mere statistical trends and reflect the functional necessities of globular proteins by amino acid properties according to a solvent-solvent (water → interior of a folded protein) partitioning model. Within this model the primary driving force is that of water-water attractions that exceed water-amino acid attractions. Hydrophobicity is not a force that repels amino acids from water, but rather that water molecules attract each other more. When hydrophobic amino acids are exposed to water, clathrate shells spontaneously form at those areas, creating an anchored aqueous patch of ordered water molecules with surface tension. Thus, the preferred hydrophobicity scale of hydrophobic proclivities as we have described here provides significant new information to alignment algorithms and in particular our TMATCH algorithm (described elsewhere), optimized to work with our hydrophobicity proclivity scale.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
https://chemrxiv.org/authors/David_Cavanaugh/8853095
https://chemrxiv.org/authors/David_Cavanaugh/8853095