Keywords
Mycobacterium tuberculosis, hemoglobin, water molecules, ligand interaction
This article is included in the Oxygen-binding and sensing proteins collection.
Mycobacterium tuberculosis, hemoglobin, water molecules, ligand interaction
Minor changes were made to a few sentences in order to be more specific and further clarify our intended meaning.
See the authors' detailed response to the review by Marco Nardini
See the authors' detailed response to the review by Michael Wilson
Mycobacterium tuberculosis, the causative agent of human tuberculosis, affects approximately two billion people world-wide, causing over three millions deaths each year1. The genome of this pathogenic organism includes two genes, glbN and glbO, which encode for two proteins, termed here truncated hemoglobin N (Mt-trHbN) and truncated hemoglobin O (Mt-trHbO), belonging to the truncated hemoglobin (trHb) family of heme proteins, widely distributed in eubacteria, cyanobacteria, microbial eukaryotes and plants2,3.
The truncated hemoglobin family exhibits a three-dimensional structure similar to the common globin fold of myoglobin, but significantly smaller. The secondary structure of trHbs consists of four α-helices arranged in a two-over-two antiparallel sandwich instead of the common three-over-three helix globin fold. Phylogenetic analysis has distinguished three different groups of truncated hemoglobins, classified as groups I, II and III, also called N, O and P, respectively.
It has been shown that group I Mt-trHbN catalyzes the detoxification of •NO in the presence of O24,5. The first step of this mechanism involves O2 migration and binding. Subsequently, •NO migrates to the active site and reacts with the heme-bound O2 to yield an unstable peroxynitrite adduct, which isomerizes to generate the relatively innocuous nitrate anion.
Several studies have examined the role of internal tunnels in ligand migration in trHbs2,5–8. Three different internal tunnels have been characterize among the trHb members, in general one or two of these tunnels is found in each protein: a long tunnel (LT) topologically positioned between helices B and E, and two short tunnels, known as the E7 Gate (E7 gate) and the short tunnel G8 (STG8), which are roughly normal to the LT, as depicted in Figure 1. The E7 tunnel corresponds to the highly conserved E7 pathway widely studied in both myoglobin and hemoglobin9–11. The STG8 tunnel is analogous to that found in Mt-trHbN, next to the key residues VG8 and IH11. Previous results indicate that WG8, an absolutely conserved residue in groups II and III truncated hemoglobins, is involved in hindering ligand migration in Mt-trHbO by blocking both STG8 and LT (Figure 1)12–14. In addition, the presence of a smaller residue at the G8 position in the Mt-trHbO mutant (WG8F) was observed to increase the small ligand association constant, although the molecular details of this process were not investigated12–14. It has also been noted that in myoglobin, M. Tuberculosis trHbN as well as in T. fusca trHbO, internal water molecules were observed to block the heme accessibility, thus delaying ligand binding15–17.

The Long Tunnel (LT) and Short Tunnel G8 (STG8) are shown in orange.
By performing CO association kinetic constant measurements (kon CO), •NO decomposition, and molecular dynamics (MD) simulations of Mt-trHbN, we addressed molecular mechanisms that control ligand association in M. tuberculosis truncated hemoglobins.
The trHbN G8 mutants (VG8W and VG8F) were prepared using the Stratagene QuikChange mutagenesis kit. The following primers were designed using Primer3 http://biotools.umassmed.edu/bioapps/primer3_www.cgi18 to generate single amino acid substitutions (underlined): i) WG8: forward primer 5’–CACTTCAGCCTGTGGGCCGGACACTTGG–3’ and reverse primer 5’–CAAGTGTCCGGCCCACAGGCTGAAGTG–3’; ii) FG8: forward primer 5’–ACCACTTCAGCCTGTTCGCCGGACACTTG–3’ and reverse primer 5’–CAAGTGTCCGGCGAACAGGCTGAAGTGGT–3’. Polymerase chain reaction (PCR) amplification of pET9b carrying the glbN gene with the aforementioned primers was conducted following manufacturer’s instructions. The PCR mix consisted of 5 µl 10x reaction buffer, 5–50 ng double stranded DNA template, 125 ng oligonucleotide primer 1, 125 ng oligonucleotide primer 2, 1 µl dNTP mix, 1 µl PfuUltra HF DNA polymerase and double distilled H2O to a final volume of 50 µl. The PCR reaction was 95°C for 30 s, followed by 16 cycles of: 95°C for 30 s, 55°C for 1 min and 68°C for 4 min. The reaction mix was then digested with DpnI to remove parental methylated DNA. Plasmid containing the mutated gene was then purified and used to transform Escherichia coli XL-1 Blue electrocompetent cells. Cells were provided by Invitrogen. Constructs were checked by sequencing.
All chemicals and reagents were obtained from Sigma Aldrich, unless indicated otherwise. The trHbN protein variants were purified using standard techniques reported for other bacterial globins19. Briefly, mutated constructs were used to transform E. coli BL21 DE3 pLysS. Starter cultures grown overnight in LB supplemented with kanamycin (50 μg ml-1) and chloramphenicol (35 μg ml-1) were used to inoculate 6 batches of 1 L LB medium at 1% (v/v), supplemented with kanamycin and 3 μM FeCl3. Once cultures reached an OD600 of around 0.4, expression of trHbN was initiated by the addition of 1 mM IPTG and grown for a further 4 h. Cells were harvested by centrifugation at 5500 rpm for 20 min at 4°C and stored overnight at -20°C. After thawing, cells were resuspended in 40 ml buffer (10 mM TRIS-HCl (pH 7.0) with 1 mM EDTA, 10 mM DTT, 45 μg ml-1 phenylmethylsulphonyl fluoride, 500 μg ml-1 RNase and 50 μl DNase), homogenized using a Douce homogeniser and ultracentrifuged at 44,000 rpm for 1 h at 4°C. The supernatant, red in color, was loaded onto a 30 ml DEAE Sepharose Fast Flow column (Pharmacia Biotech) equilibrated with 10 mM TRIS-HCl (pH 7.0), washed with the same buffer until the UV trace returned to baseline, and eluted via a gradient from 0 to 1 M NaCl in 10 mM TRIS-HCl using an Akta purifier (GE Healthcare Bio-Sciences, Amersham Biosciences, U.K. Ltd.). Fractions that were most red in color were concentrated using a Vivaspin 20 concentrator (Sartorius Stedim Biotech) to around 5 ml and loaded onto a gel filtration Superdex 75 column, equilibrated with 0.15 M NaCl in 10 mM TRIS-HCl (pH 7.5); again, fractions with the most color were collected, combined and stored at -80°C. Purity was checked using gel electrophoresis and analysis of the heme-to-protein ratio (410 nm and 280 nm in the UV-visible absorption spectrum).
Rapid mixing experiments were conducted with a thermostated stopped flow apparatus (BioLogic SFM-300). Kinetics of carbon monoxide (CO) binding to determine the kon CO were measured on the deoxy state of mutant and wild type globins at 20ºC. Solutions containing 5 μM protein in a 100 mM sodium phosphate at pH 7.0 were degassed in a nitrogen atmosphere and reduced with an equimolar concentration of sodium dithionite and mixed with increasing CO concentrations. The observed pseudo first-order rate constant (kobs) was determined by fitting the absorbance decay resulting from association of the protein with CO, to a single exponential function. Kinetic rate constants (kon CO) were obtained from the slope of the plots of kobs as a function of CO concentration.
To determine rates of nitric oxide (•NO) decomposition by wild type and mutant Mt-trHbN proteins, •NO was added, as ProliNONOate, to a solution of 50 mM KPi buffer with 50 μM EDTA (pH 7.5), 100 μM NADPH and 100 nM E. coli ferredoxin reductase inside a thermoregulated, magnetically stirred reaction vessel. Mt-trHbN (2 μM) was added at the apex of the signal response to 2 μM ProliNONOate and •NO decay was followed until depleted using an •NO electrode (World Precision Instruments). Rates of •NO decay were calculated for each protein by determining the time taken for peak •NO concentrations to decay by 0.5 μM and were expressed per μM heme, determined spectrally by the peak in the Soret region at 410 nm.
The starting structure corresponds to Mt-trHbN crystal structure (PDB entry 1IDR http://www.rcsb.org/pdb/explore/explore.do?structureId=1IDR), at 1.9 Å of resolution20. The protonation state of the amino acids was assumed based on the environment of the residues in the crystal structure. All solvent-exposed His residues were protonated at the N-δ atom, as well as the proximal HisF8, because of its coordination to the heme iron. An octahedral box of 10 Å of radius, which corresponds to 5234 explicit water molecules was added to the system. TIP3P water molecules were used by tLEaP module of the AMBER12 package21. The param99 Amber force field was used for all the aminoacid parameters22 except heme parameters which were developed in our group23 and strongly validated for being used in several studies of heme proteins24–30. Periodic boundary conditions were used for all the simulations performed with a 9 Å cutoff. Particle mesh Ewald (PME) summation method for treating the electrostatic interactions. The SHAKE algorithm was used to keep constant the non-polar hydrogen equilibrium distance. Temperature and pressure were kept constant with Langevin thermostat and barostat, respectively, as implemented in the AMBER12 program21. The equilibration simulation protocol was performed as follow: (i) slowly heating the system from 0 to 300K for 20 ps at constant volume, by using harmonic restraints of 80 kcal/mol A2 for all Cα atoms and (ii) pressure equilibration of the whole system during 1 ns at 300K with restrained atoms in (i). (iii) Unconstrained 100 ns molecular dynamics simulation at constant temperature (300K) was performed.
In silico mutant proteins were built by using tLEaP module of AMBER12 package21, and underwent the same protocol used for wild type protein. Root Mean Square Deviation (RMSD) was used as structure stability controls. All structures were observed to be stables during the time scale of the simulation (Figure S1).
The free energy profile for the CO migration process inside the protein tunnel/cavity system was computed by the Implicit Ligand Sampling (ILS) approach that post-processes, using a probe molecule, an MD simulation performed in the absence of the ligand. This method was thoroughly tested for heme proteins32. ILS calculations were performed on a rectangular grid (0.5 Å resolution) that includes the whole simulation box (i.e. protein and the solvent) and the probe used was a CO molecule. Calculations were performed on 5000 frames taken from the last 90 ns of simulation time. The values for grid size, resolution and frame numbers were tested in a previous study32. Analysis of the ILS data was performed using an ad-hoc fortran-90 program available upon request32. Moreover, ILS has been shown to yield quantitative results for ligand migration processes when compared with more costly free energy methods that treat the ligand explicitly.
Although CO is not the natural ligand of the hemeproteins, it is widely used as a probe for ligand association studies due to its ease of use. In order to address the molecular determinants controlling ligand migration we performed CO ligand association constant measurements of wild type Mt-trHbN and two mutants: VG8F and VG8W. Kinetic traces for CO binding were measured through the absorption changes at the CO adduct peak position (λ=423 nm; Figure 2). Association of CO is well described by a single exponential decay, whose rate constant (kobs) depends linearly on CO concentration and the slope can be interpreted as kon CO. A significant kon CO decrease for VG8F (715 ± 27 mM-1s-1), and an even larger decrease for VG8W (48 ± 1 mM-1s-1) was observed in relation to that observed for the wild type protein (4495 ± 357 mM-1s-1) (Figure 3). Table 1 summarizes the measured kon CO values for wild type and mutant Mt-trHbs O and N, and is presented alongside literature data.
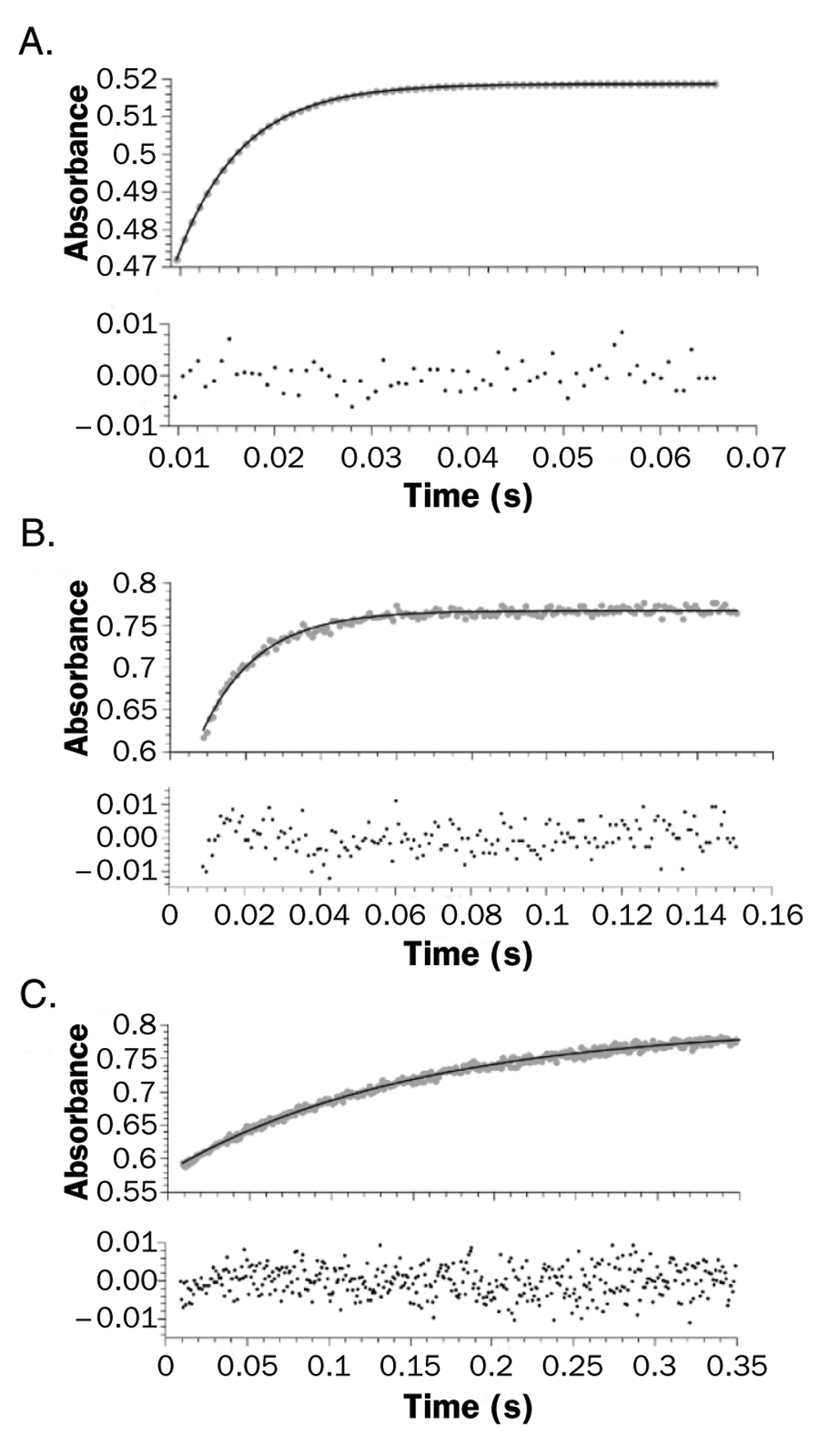
Stopped-flow time course for the reaction of reduced 5 μM wild type (A), VG8F (B), and VG8W (C) mutants in 100 mM phosphate buffer at pH 7. The reaction was monitored at 423 nm (grey dots) and the line shows the best first order fit.
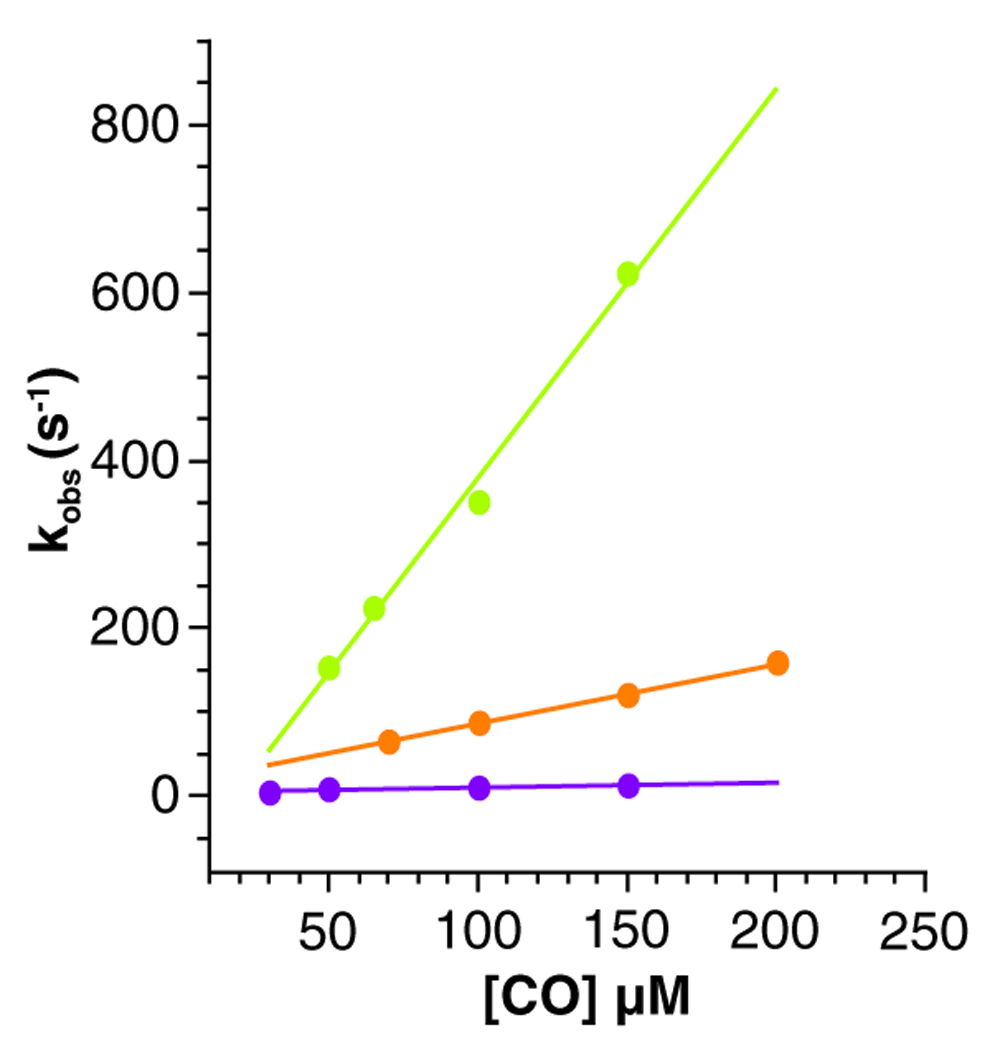
Curves for wild type (green), VG8F (orange) and VG8W (violet) mutants as a function of CO concentration in stopped flow measurements are shown. The time courses are measured at different CO concentrations ranging from 10 to 200 µM (after mixing). Continuous line corresponds to linear fit of kobs rates.
Small ligand association in the trHb family is presumably regulated by two main processes: i) ligand migration from solvent bulk to the protein distal site cavity, ii) displacement of water molecules from the distal site cavity15–17,35. With this in mind, we performed classical MD simulations as they allow us to investigate both processes involved in ligand association. Ligand migration was studied using ILS calculations for the wild type, as well as both VG8F and VG8W mutant proteins. Displacement of retained water molecules in the distal site was considered by performing classical MD simulations and analyzing the solvation structure in each active site.
The wild type Mt-trHbN presents two tunnels available for ligand migration, the LT and the STG8 (Figure 4A). On the one hand, the LT connects three internal cavities: (trHb : CO)1, (trHb : CO)2 and (trHb : CO)3. The STG8, on the other hand, has the distal site cavity (trHb : CO)1 connected to both the cavity (trHb : CO)2 and the solvent, although the cavity (trHb : CO)2 plays only a secondary role, due to the fact that it does not alter the energy migration profile along the STG8 straight from the distal cavity to the solvent. The VG8F mutant conserves both tunnels, although they are constrained compared to those in the wild type. In the VG8W case, however, the energy profiles suggest a completely blocked STG8 and a LT for which the accessibility to the iron heme is partially reduced.

(A) Schematic representations of the residues involved in the heme distal site and tunnels, the two tunnels and cavities estimated with ILS for the wild type form. Free energy profiles over STG8 (B) and LT (C) connecting the solvent with the distal site through the cavities (trHb : CO)1, (trHb : CO)2 and (trHb : CO)3, for wild type (green), VG8F (orange) and VG8W (violet) mutant Mt-trHbN. Circles represent calculated free energy values with the ILS method and lines correspond to a fitting estimation of these calculated values. The x coordinate represents the Fe-CO distance along the pathways.
In order to quantify the contribution of the single G8 mutation we computed free energy profiles for CO migration through both LT and STG8 tunnels (Figure 4B, 4C). The free energy was set to a value of 0 kcal/mol where CO ligand is fully solvated- at 13 Å and 24 Å from the Fe atom, for STG8 and LT respectively. Wild type Mt-trHbN presents small barriers (~2 kcal mol-1) for CO migration from the solvent to the active site cavity (trHb : CO)1 through both tunnels.
The active site water molecules occupancy was computed for all three systems by performing 200 ns of MD simulations with explicit water molecules. In each case a water molecule was able to access the active site and was stabilized by the iron and the distal site residues (Figure 5). Specifically, in both wild type and VG8F Mt-trHbN a water molecule was present for approximately 40% of the length of the simulation (Figure 5A, 5B). The VG8W mutant active site, on the other hand, is occupied by water molecules in 80% of the simulation time, probably due to the hydrogen bonding capacity of W (Figure 5C).

(A) wild type, (B) VG8F and (C) VG8W forms showing, on the basis of MD simulations, the hydrogen-bond network (dotted lines) stabilizing a water molecule above the iron heme. The percentages depicted as insets in the figure correspond to active site water occupancy during MD simulation.
Mt-trHbN has previously been described as a dioxygenase, capable of O2-dependent •NO consumption36,37. Consequently, •NO decomposition by purified Mt-trHbN and the VG8F, VG8W mutants was determined in a reaction mixture containing buffer, NADPH and E. coli FdR, to enable cyclic restoration of heme iron to the oxyferrous state. Figure 6A shows that in the absence of protein (red trace) decay of the •NO signal was monophasic until •NO was exhausted. The decay of NO in the presence of Mt-trHbN (black trace) was biphasic, with an almost linear initial rapid rate in decay, which was used to compare the various Mt-trHbN derivatives, followed by a slower rate in decay. This suggests that •NO is not being turned over in a cyclic manner, but is simply binding available heme. The chemical step being measured in this assay is the reaction between •NO and the oxyferrous heme; once this reaction has concluded, we assume that the heme is restored from ferric back to ferrous. We are unsure why the reaction is single turnover but it could be due to (a) rapid binding of •NO to the ferrous complex before oxygen can bind, rendering it unable to bind oxygen and initiate the reaction or (b) due to slow reduction of Mt-trHbN by the non-native E. coli FdR. •NO consumption results show that the VG8F and VG8W mutants have a statistically significant reduced •NO binding capacity compared to HbN (Figure 6B).

(A) •NO decay was monitored amperometrically in the absence (red trace) and the presence (black trace) of Mt-trHbN added at the apex of the signal response to 2 μM ProliNONOate. Data are representative of 3 technical repeats. (B) Mean rates of •NO decay in the presence of wild type Mt-trHbN or site-directed mutants from 3 technical repeats ± S.E.M * P < 0.05, unpaired t-test.
CO association kinetic constant measurements as well as MD simulations of Mt-trHbN wild type and site-specific mutants were performed to analyze the role of tunnels and water molecules in the ligand association process. ILS calculations showed that the main tunnels of wild type Mt-trHbN, STG8 and LT, were partially blocked in the VG8F mutant and STG8 was nearly completely blocked in the VG8W mutant. The analysis of water molecules showed that VG8W increases the probability of the presence of a water molecule in the distal site, which may interfere with the association process. Consistently, the association kinetic constants of CO for both Mt-trHbN mutants showed a decrease of slightly less than one order of magnitude when VG8 is replaced with F and two orders of magnitude when VG8 is replaced with W. Moreover, our data also showed that both mutants have less capacity of •NO binding than wild type Mt-trHbN.
Interestingly, the Mt-trHbN VG8W mutant presents a similar kon CO to the wild type Mt-trHbO, showing that a single residue is responsible for the differential accessibility in these proteins. The results support the idea that STG8 and LT are the main channels for CO migration in the deoxygenated Mt-trHbN, as blocking these tunnels decreases the ability of CO to access the heme pocket. Although in both the mutated Mt-trHbN and wild type Mt-trHbO the STG8 is blocked by WG8, the LT remains open in Mt-trHbN, allowing CO access into the heme cavity, whereas the main tunnel for CO migration in Mt-trHbO is the E7, as was previously described7. This fact shows that although the kon CO from mutant trHbN and wild type trHbO members are very similar, the ligand enters through different pathways, evidencing the complexity of mechanisms that regulate the ligand association process in these proteins.
F1000Research: Dataset 1. Experimental and theoretical calculations raw data, 10.5256/f1000research.5921.d4209139
IB, LB, SC, RR, KLD, LAHB designed and performed the experiments. JPB, LB, MAM, DAE designed and analyzed the MD simulations. DAE, MAM, KSD and LB provided expertise in the field. KD, RLP, KLD, SS provided the experimental sample. JPB and LB wrote the manuscript. KSD, SS, LAHB, MTT and RP contributed to the experimental design and preparation of the manuscript. All authors were involved in the revision of the draft manuscript and have agreed to the final content.
This work was supported by Framework program 7 NOstress Grant, CONICET, University of Buenos Aires, and Agencia Nacional de Promoción Científica y Tecnológica, National Institutes of Health Grant R01AI095173 and Universidad de la República (CSIC, Uruguay) to R. R. Additional funding to SC and RR was provided by PEDECIBA (Progama de Desarrollo de Ciencias Básicas, Uruguay) and CeBEM (Centro de Biología Estructural del Mercosur). IB and JPB hold CONICET PhD fellowships. LB is a Pew Latin American Fellow. LB, DAE and MAM are members of CONICET.
I confirm that the funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
FJ Luque is acknowledged for useful discussions and suggestions. Mehrnoosh Arrar is acknowledged for close reading of the manuscript.
| Views | Downloads | |
|---|---|---|
| F1000Research | - | - |
|
PubMed Central
Data from PMC are received and updated monthly.
|
- | - |
Click here to access the data.
Spreadsheet data files may not format correctly if your computer is using different default delimiters (symbols used to separate values into separate cells) - a spreadsheet created in one region is sometimes misinterpreted by computers in other regions. You can change the regional settings on your computer so that the spreadsheet can be interpreted correctly.
Provide sufficient details of any financial or non-financial competing interests to enable users to assess whether your comments might lead a reasonable person to question your impartiality. Consider the following examples, but note that this is not an exhaustive list:
Sign up for content alerts and receive a weekly or monthly email with all newly published articles
Already registered? Sign in
The email address should be the one you originally registered with F1000.
You registered with F1000 via Google, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Google account password, please click here.
You registered with F1000 via Facebook, so we cannot reset your password.
To sign in, please click here.
If you still need help with your Facebook account password, please click here.
If your email address is registered with us, we will email you instructions to reset your password.
If you think you should have received this email but it has not arrived, please check your spam filters and/or contact for further assistance.
Comments on this article Comments (0)